VIENNA - U.K. researchers have developed an accurate matching algorithm to deal with the gnarly problem of integrating prone and supine virtual colonoscopy datasets, according to a presentation at ECR 2013.
The time-consuming task must be completed for each virtual colonoscopy (also known as CT colonography, or CTC) exam with findings that require confirmation, but successful automation and some tweaks to speed up the process could improve radiologists' workflow.
The researchers tested their method by applying it to 17 CTC cases -- including several containing collapsed lumen segments -- and achieved a high fold-matching accuracy of 96%. The method significantly improved cylindrical registration as well, achieving a mean error of 6 mm from the actual location measured at 1,743 reference points.
"At an accuracy of 96.1%, the algorithm performs comparably to the human reader," said Dr. Thomas Hampshire, from the Centre for Medical Image Computing at University College London. "There is also a similar level of performance in fully connected cases in comparison to cases exhibiting one or more areas of luminal collapse."
CTC is a highly accurate technique for detecting colorectal polyps and cancer, and the acquisition of both prone and supine CT datasets allows the radiologist to distinguish between fixed colonic pathology and mobile fecal residue. However, doing this accurately requires matching the locations of polyp candidates on both prone and supine datasets, a task that can be difficult and time-consuming due to the significant deformations of the colon that occur when the patient moves from one position to the other during the scan.
The new technique differs from previous methods that were based on the colon centerline, which does not always work well in patients with collapsed lumen segments.
"Popular current tools are based on matching centerlines, whereas our method has the advantage of matching corresponding locations on the endoluminal colon surface," Hampshire explained.
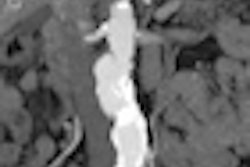
 |
| With the new technique, locations of fold matches are superimposed on the unfolded prone image, along with the amount of displacement to the corresponding folds in the supine image. All images courtesy of Dr. Thomas Hampshire. |
How it works
"We can begin by mathematical simplification to unfold these rigid structures into a more simple 2D cylindrical form, and we can then find correspondence between these two cylinders by first extracting and matching haustral folds, and then refining the registration using an elementary shape index which is a measure of the surface shape," he said. Applying this index, polyps would have a value of about 1, versus about 0.75 for haustral folds.
Next, a segmentation scheme is applied over the colonic surface mesh, which results in a smooth labeling of the entire colonic surface as either a fold or nonfold, he said. Prone and supine positions can now be matched to a location on each dataset.
For efficient labeling of the folds, a unary cost function is applied to match the depth map data, one fold pair at a time, between the supine and prone positions. To accomplish this, virtual cameras are focused on the haustral folds to be matched, depth map images are rendered at each camera location, and then the camera positions are optimized so that the two datasets match each other more closely, Hampshire said.
An additional pair-wise function compares the geometric relationship between pairs of haustral folds in the prone and supine images, and a Markov random field is used to estimate the correct fold labeling. This process allows for landmark correspondence of positions in two views even when endoluminal collapses occur, which is common in clinical practice, he said.
Using unfolded cylindrical representations of the colon surface, the prone image is then deformed to more closely match the supine image. A second intensity-based registration process then matches the fold locations more precisely.
Nearly equivalent to readers
To validate the method, Hampshire and colleagues compared the accuracy of the algorithm against known correspondences in the reference standard -- and against the accuracy of three individual readers against the reference standard. The algorithm accuracy was similar to the reader accuracy at 96.1%.
 |
Finally, the group compared accuracy of registration over the entire length of the colon, using all 1,743 corresponding fold pairs contained in the reference standard. The results were slightly better for fully distended colons versus those containing collapsed segments, demonstrating a mean overall mean error of 6 mm.
 |
Unfortunately, long processing times are a drawback of the method in its current research phase, Hampshire told AuntMinnie.com in an email. "We are currently working with research code which is in the region of a few hours" per dataset, he noted. "We are optimizing the entire pipeline over the next six months to develop a clinical prototype, and hope to get the processing time to 10 to 20 minutes so that it can fit into the clinical workflow."