Biliary Atresia and Hepatobiliary Scintigraphy in Infancy:.
Normal hepatobiliary exam in infants:
In neonates, extraction of the radiotracer by the liver is normally prompt with a uniform distribution. Maximum tracer accumulation is reached within 5 minutes. The gallbladder can be seen as early as 10 minutes, but occasionally it is not seen in the neonatal period (possibly related to biliary stasis or reduced bile flow). Bowel activity is usually seen by 30-40 minutes. The hepatic and common bile ducts are not normally visualized in the neonatal period. The normal hepatic extraction fraction is greater than 92% and the hepatic clearance T1/2 is normally less than 37 minutes. [1]
Hyperbilirubinemia:
Hyperbilirubinemia in the neonatal period is common and is typically the result of benign physiologic jaundice. Potential causes of cholestasis in neonates inlcude viral infection, TPN, metabolic disorders, choledochal cyst, and biliary atresia [7]. If direct hyperbilirubinemia persists beyond 2 weeks of age in a full-term infant, however, further evaluation is necessary to exclude an underlying pathologic abnormality.
Exam Technique for the evaluation of hyperbilirubinemia: Tc-Mebrofenin (trimethylbromo-IDA) is administered according to body weight with a minimum dose of 30 MBq (0.81 mCi) and a maximum dose of 185 MBq (5 mCi). Mebrofenin has a hepatic extraction of 98% and urinary excretion of 1.5%. It is the best agent to use with high bilirubin levels due to it's high resistance to displacement by bilirubin. Hepatocyte uptake is greater than 70% even with bilirubin levels greater than 20 mg/dl. Fasting for a minimum of 3 hours before the exam is recommended.
Differential considerations for neonatal hyperbilirubinemia include:
From: J Nucl Med 1998; Howman-Giles R, et al. Hepatobiliary scintigraphy in infancy. 39: 311-319
- Physiologic jaundice
- Biliary atresia
- Neonatal hepatitis: CMV, Hepatitis A, Hepatitis B, Rubella, Toxoplasmosis
- Choledochal cyst
- Bile plug syndrome
- Bile duct paucity syndrome (Alagille syndrome)
- Cystic Fibrosis
- Total parenteral nutrition (due to cholestasis)
- Gallstones (Risk factors for cholelithiasis in the neonatal age group include prematurity, prolonged fasting, sepsis, blood transfusions, the use of diuretics, narcotic analgesics, and prolonged hyperalimentation)
- Metabolic disorders: Tyrosinemia, Wolmans disease, Galactosemia, and Fructosemia
- Byler disease: A rare disorder of bile excretion that causes severe cholestasis
Biliary Atresia:
Clinical:
Biliary atresia is a progressive oblietrative cholangiopathy that
eventually leads to liver cirrhosis [7] and it is the most common
fatal liver disorder in children. It typically presents by 1-2
months of age and affects males and female equally. Persistent
direct hyperbilirubinemia and jaundice are the usual symptoms.
Most patients with biliary atresia are born at full term, have a
normal birth weight, and initially seem healthy [9]. Associated
anomalies are found in 10-25% of patients and include polysplenia,
preduodenal or absent portal vein, anomalous hepatic artery,
intestinal malrotation, congenital heart disease, Trisomy 18, and
choledochal cyst. In contrast, prematurity is a risk factor for
neonatal hepatitis and often necessitates the use of prolonged
parenteral nutrition which leads to cholestasis [9].
If untreated, all patients develop fatal biliary cirrhosis by age 2 to 3 months. The Kasai procedure (portoenterostomy) is performed initially to re-establish bile flow and it can reduce or delay the need for liver transplant during infantcy. Biliary flow can be reestablished in 90% of children if surgery is performed before the infant is 10 weeks old. The success rate drops to 20% if surgery is delayed more than 3 months. Nonetheless, many treated cases progress to biliary cirrhosis. Long term survival occurs in only 16% of cases, and only about 8% remain jaundice free with normal liver function tests. Cholangitis is also common in patients with biliary atresia and it occurs in more than half of all patients who have a portoenterostomy. Liver transplantation is often required in patients with biliary atresia.
Biliary atresia is a developmental anomaly (atresia/fibrosis) of extrahepatic ducts with variable, generally progressive, involvement of the intrahepatic ducts and gallbladder. It is felt to be secondary to an intrauterine insult during hepatic duct development- possibly an obliterative cholangitis (?rheovirus type 3). Histologically, there is periportal fibrosis, proliferation of small intrahepatic bile ducts, and focal or total absence of extrahepatic ducts.
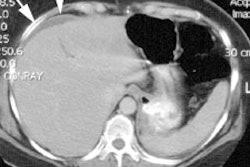
The presence of biliary cysts has been documented by MRI in patients with biliary atresia [2]. Cysts are noted in 5 to 25% of patients. The cysts can be multilocular (located in the porta hepatis) or unilocular (which can be multiple and can be found in the porta or in the liver). The presence of cysts is associated with a history of clinical cholangitis (50% of cases).
X-ray:
Sonography: On ultrasound non-visualization of the
gallbladder is a very specific finding for biliary atresia [5].
Gallbladder visualization, however, does not exclude the diagnosis
of biliary atresia.
Other findings seen in biliary atresia include:
1- The "triangular cord" sign- representing a focal area of
increased echogenicity anterior to the bifurcation of the portal
vein (anterior to the right portal vein [6]) measuring greater
than 4 mm that represents the fibrotic remnant of the extrahepatic
biliary tree (sensitivity 43-80%, specificity 73-100% [6,7]-
therefore absence of a triangular cord sign does not exclude
biliary atresia [7]). However, a meta-analysis reported a pooled
sensitivity of 85% and a specificity of 95% [9].
2- Gallbladder abnormalities- length less than 15-19 mm, irregular or lobular gallbladder wall, and/or indistinct mucosal lining ("gallbladder ghost" triad); and 3- absent common bile duct [5,6]. Intrahepatic biliary dilatation is not typically seen as these ducts are also diseased (fibrosed).
Scintigraphy:
Phenobarbital priming (5mg/kg/day in 2 divided doses) is
instituted for 5 days prior to the exam and patients should have a
serum phenobarbitol level of more than 15 ug/mL [8]. Phenobarb is
a potent inducer of the liver microsomal enzyme system, increases
bilirubin conjugation, and provides better excretion of the
compound for earlier identification of a patent biliary tree [3].
Phenobarbital pretreatment improves the specificity of the
examination from 63 to 94% [1]. However, other series have
reported a lower specificity for the phenobarbitol examination (as
low as 43%) and have questioned whether phenobarbitol priming is
necessary prior to imaging patients with suspected biliary atresia
[3]. Ursodeoxycholic acid (UDCA) is a bile acid used in the
treatment of cholestatic liver diseases in children [3]. Priming
with UDCA requires only 2- days and can also improve the
specificity of the exam [3].
Normal uptake and excretion of the tracer in a jaundiced
neonate excludes the diagnosis of biliary atresia. In
biliary atresia there is usually normal prompt clearance of tracer
from the blood and normal hepatic concentration with a high liver
to heart ratio at 5 minutes (and normal hepatic extraction
fraction > 92%). Subsequently, there is NO EXCRETION from the
liver with non-visualization of the biliary tree and bowel at 1,
4, and 24 hours. There have been case reports of patients with
biliary atresia in which bowel activity was seen in the early
neonatal period, but not on subsequent exams (likely due to the
progressive nature of the obliteration of the bile ducts [1].
Beyond three months of age, the prolonged obstruction will begin
to affect liver function and hepatic extraction of the tracer will
deteriorate making differentiation from hepatitis more difficult.
Increasing the dose of tracer administered to 2 mCi will improve
count statistics on 24 hour delayed images. In the evaluation of
biliary atresia, hepatobiliary scintigraphy has a sensitivity of
97-100%, specificity of 70-94%, and accuracy of 91% [1,9].
However, other authors report a lower sensitivity of between
75-80% [3,8] and a false positive rate of 13% [9]. None-the-less,
the negative predictive value is very high and approaches 100%
[8].
|
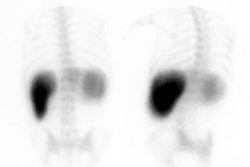
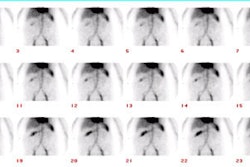
Biliary atresia: The patient presented for evaluation of suspected biliary atresia at 3 months of age. The exam demonstrated delayed, but homogeneous uptake of tracer on early images (click early image to enlarge). No bowel activity can be seen. Delayed images at 24 hours demonstrated persistent hepatic activity, no bowel activity, and bladder activity secondary urinary excretion of the tracer. A shielded image was also obtained to enhance detection of any subtle GI activity. The patient was confirmed to have biliary atresia. |
|
|


Following portoenterostomy there is usually good extraction of the tracer by the liver with prompt excretion into the surgical diversion. In patients that developing a complicating cholangitis, extraction of tracer will remain moderately good, but there will be delayed excretion. There may be irregular distribution of the tracer with focal areas of holdup in the intrahepatic ducts.
Neonatal Hepatitis:
Scintigraphy: In neonatal hepatitis, depending on the severity of cholestasis and hepatocellular dysfunction, different scan patterns may be noted. Typically, patients with neonatal hepatitis will demonstrate poor hepatic tracer uptake due to hepatocellular dysfunction (decreased hepatic extraction fraction), prolonged blood pool activity, poor biliary excretion, delayed transit into the bowel, and renal excretion. The time-activity curve typically demonstrates an early peak due to vascular activity and subsequently parallels a curve of the cardiac blood pool because there is minimal active uptake by the liver. If the hepatocellular dysfunction is severe, there may be no evidence of bowel activity even at 24 hours. SPECT images may aid in detecting small amount of bowel activity not apparent on planar images [1]. Phenobarbital premedication will enhance hepatic excretion in patients with neonatal hepatitis.
Choledochal Cyst:
Clinical:
A choledochal cyst is a congenital focal cystic or fusiform dilatation of the common bile duct with or without associated proximal intrahepatic ductal dilatation. An anomalous junction of the pancreatic and biliary ducts is seen in 95% of cases and is felt to be related to an increased incidence of pancreatitis in these patients. Patients with choledochal cysts usually present prior to the age of 10 years and females are affected more than males (4:1). Symptoms include pain (90%), intermittent jaundice (33%), RUQ mass (25%), choledocholithiasis (50%), pancreatitis (33%), and cholangitis. Affected patients are at an increased risk of developing cholangiocarcinoma (10% of patients). Treatment consists of early surgical excision or anastamosis to the intestine in order to prevent biliary cirrhosis. Complications include: Cholangitis, pancreatitis, stone formation, and cholangiocarcinoma (20x's general population risk). Choledochal cysts may be subclassified as:
Type I: Commonly associated with an anomalous pancreaticobiliary ductal system (ie: a common junction of CBD and pancreatic duct within the sphincter of Oddi).
- 1A: Cystic dilatation of the common bile duct with marked dilatation of part or all of the extrahepatic biliary tree and a normal intrahepatic biliary tree. This form occurs in 80-90% of cases [1].
- 1B: Focal segmental dilatation of the CBD
- 1C: Fusiform dilatation of the common bile duct and common hepatic duct with a normal intrahepatic biliary tree
Type II: Diverticular outpouching from CBD or CHD, it accounts for only 3% of cases.
Type III: Choledochocele: Represents a cystic dilatation of the intramural duodenal segment of the CBD in the region of the ampulla of Vater. The wall of the common bile duct herniates into the duodenum. This may occur secondary to a congenital stenosis of the ductal orifice or be acquired, possibly secondary to stone passage with stenosis and inflammation. On UGI exam, a choledochocele appears as a well defined, smooth filling defect on the medial wall of the descending duodenum.
Type IV:
- 4A: Multiple intra and extrahepatic cysts
- 4B: Multiple extrahepatic cysts
Type V: Caroli's disease- dilatation of one or several segments of the intrahepatic bile ducts without dilatation of the common bile duct.
X-ray:
Scintigraphy: A variety of appearances can be seen on the scintigraphic exam. Uptake of the radiotracer by the liver is usually normal. Initially the bile filled cyst may appear as a photopenic area, but on delayed images the cyst will fill with tracer and appear as a hot spot. If the cyst is obstructive, there may be non-visualization of the CBD and bowel activity may not be identified.
Other imaging modalities: Sonography will demonstrate a cystic mass in the porta separate from the gallbladder (which may contain sludge or stones). A dilated common duct/common hepatic duct can be seen entering the mass. The intrahepatic ducts are generally not dilated, although this can be seen. On UGI there may be anteroinferior displacement of the gastric antrum or compression of the duodenal bulb if the cyst is large. On cholangiography the cyst will be shown to communicate with the biliary tree and may be filled with stones or contain a carcinoma.



Bile-Plug Syndrome:
The disorder affects neonates and is associated with severe hemolytic disease (Rh or ABO incompatibility), dehydration, total parenteral nutrition, and after extensive ileal resection surgery. A viscous bile leads to extrahepatic obstruction and calculi may be seen. [1]
Scintigraphy: Scintigraphic findings are variable. Usually there is good extraction of the tracer by the liver with poor excretion (typical of severe cholestasis). Occasionally there is poor tracer uptake due to hepatocellular dysfunction, and rarely, no excretion may be seen- making differentiation from biliary atresia impossible. There is usually intrahepatic biliary dilatation identified on ultrasound and the plug may be visualized
Bile Duct Paucity Syndrome:
Bile duct paucity syndrome is a rare disorder secondary to the defective development of intrahepatic bile ducts. It includes a number of syndromes, the most common of which is Alagille syndrome (or Arteriohepatic dysplasia). Alagilles is an autosomal dominant disorder. Associated anomalies include peripheral pulmonic stenosis, tetrology of Falot or complex congenital heart disease, distinct facies, vertebral anomalies (spina bifida or butterfly vertebral bodies), and cholestasis. There is no effective surgical treatment. [1]
Scintigraphy: On hepatobiliary imaging there is usually good hepatic uptake of the tracer, with poor excretion and minimal gut activity. Occasionally there is no excretion and differentiation from biliary atresia cannot be made.
Cystic Fibrosis:
Clinical:
Infants with cystic fibrosis may experience mild cholestatic jaundice to severe cholestasis. Biliary obstruction can occur due to inspissation of biliary secretion and generally resolves by 3 to 4 months of age.
Scintigraphy: Hepatobiliary exams usually demonstrate moderately good tracer extraction with poor excretion. In severe cases, there may be no evidence of gut activity. Premedication with phenobarbital is highly recommended.
REFERENCES:
(1) J Nucl Med 1998; Howman-Giles R, et al. Hepatobiliary
scintigraphy in infancy. 39: 311-319
(2) AJR 1993; Jan.: p.167
(3) J Nucl Med 2004; Poddar U, et al. Ursodeoxycholic acid-augmented hepatobiliary scintigraphy in the evaluation of neonatal jaundice. 45: 1488-1492
(4) Radiology 2005; Ryeom HK, et al. Biliary atresia: feasibility of mangafodipir trisodium-enhanced MR cholangiography for evaluation. 235: 250-258
(5) Radiology 2007; Humphrey TM, Stringer MD. Biliary atresia: US diagnosis. 244: 845-851
(6) Radiology 2009; Lee MS, et al. Biliary atresia: color doppler
US findings in neonates and infants. 252: 282-289
(7) Radiology 2011; Lee SY, et al. Efficacy of US-guided
percutaneous cholecystocholangiography for the early exclusion and
type determination of biliary atresia. 261: 916-922
(8) J Nucl Med 2014; Ziessman HA. Hepatobiliary scinigraphy in
2014. 55: 967-975
(9) Radiology 2018; Kim JR, et al. Risk estimation for biliary
atresia in patients with neonatal cholestasis: development of
validation of a risk score. 288: 262-269