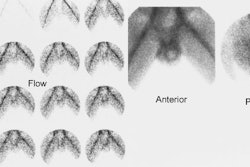
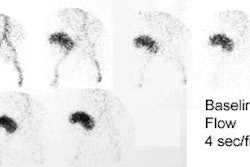
Adrenal Imaging
Meta-iodobenzylguanidine
(MIBG)
Pharmacology &
Physiology
Structurally, MIBG resembles norepinephrine and guanethidine (a neurosecretory granule depleting agent). MIBG images the adrenal medulla (NOT the adrenal cortex) and sympathetic nervous tissue. It can be labeled to either 123I or 131I. If using 123I, a 7 fold larger dose is permitted and this allows for SPECT imaging. The higher dose provides a 10 fold higher photon flux at 24 hours, and is still 3 times higher at 48 hours. The 159 keV photon of 123I is also more easily collimated and the images obtained are of higher quality. 123I images also have improved spatial resolution.
MIBG localizes to storage granules in adrenergic tissue of neural crest origin and will concentrate in catecholamine producing adrenal medullary tumors [24]. Its uptake appears to occur through the energy requiring, sodium dependent (active transport) Type I amine uptake mechanism and there is some non-energy dependent diffuse (Type II) [12]. Once inside the cell, MIBG is concentrated in the intracellular hormone storage vesicles by an energy dependent, tetrebenazine (reserpine)-sensitive mechanism [1]. Uptake is proportional to the number of neurosecretory granules within the tumor. In neuroblastoma, the agent remains within the cellular cytoplasm, free of granular storage. It's retention in neuroblastoma is related to the rapid re-uptake of agent that has escaped the cell [12].
In childhood, extra-adrenal chromaffin tissue is abundant. The largest mass of such tissue is the Organ of Z?ckerkandl which is located anteriorly below the origin of the IMA. After the age of 3 years, these extra-adrenal sites regress except in sympathetic ganglia and the carotid bodies.
Normal
distribution of MIBG:
- Adrenal medulla
131I: The adrenals are
usually not seen, but may be faintly visualized in 10-20% on
delayed images. The larger the dose of tracer used, the more
likely the normal adrenal gland will be visualized. Also, due to
the improved specific activity of MIBG used for imaging,
increased visualization of the normal adrenal gland has been
noted (up to 73% of patients on delayed imaging) [1]. The
intensity of uptake in the normal adrenal should be less than
the liver.
123I: Adrenals are seen in 30% of patients. Although the activity is generally mild (less
than liver intensity) and symmetric, it can be unilateral or
asymmetric in up to 50% of cases. Confusion can then
occur in differentiating this uptake from a pheochromocytoma. Unilateral uptake less
than or equal to the liver can represent pheochromocytoma
in 10% and 33% of cases, respectively. Unfortunately, 31% of
normal adrenals are also visualized with uptakes less than or
equal to the liver. The absence of an anatomic lesion on the
abnormal side can be helpful in excluding a tumor. In general,
most pheochromocytomas demonstrate
uptake more intense than the liver (80%). [2]
- Liver: The liver
is a major site of catecholamine degradation [13]. Liver
uptake is inversely related to catecholamine levels.
- Spleen: Probably a
manifestation of sympathetic innervation
[13]
- Urinary bladder:
Due to renal excretion of the tracer
- Colon/Bowel:
Activity is seen in up to 15-20% of patients
- Salivary glands:
Due to rich sympathetic innervation
(occasionally lacrimal gland activity may be observed
[41])
- Nasal mucosa [41]
- Heart/myocardium:
Uptake is inversely related to catecholamine levels
- Lung
- Thyroid: May be
seen in a small number of patients, even if it is being
blocked.
- Uterine and neck
muscle uptake [3]
- Upper thorax in
children [4] due to the presence of sympathetically
innervated brown fat [19]
Excretion
Most MIBG (about 85-90%) is excreted unchanged in the urine by 96 hours post injection. There is variable excretion into the gut (1 to 4%). The whole body half-life is about 24 hours, but the agent is retained in sympathetic nervous tissue for a prolonged period of time.
Indications
MIBG is used primarily to image Pheochromocytomas (Paragangliomas) and Neuroblastoma. Other APUD cell line tumors-- e.g. carcinoid, and medullary thyroid carcinoma-- demonstrate uptake of the tracer less frequently. Adrenal medullary hyperplasia may also be imaged.
Technique
Dose: 500 uCi 131I or
3 to 10 mCi 123I.
Imaging is performed at 1 and 3 days following 131I
administration, and 6 and 24 hours after 123I
administration. Thyroid uptake of the tracer and free I-131 is
blocked using Lugol's solution or
Super-saturated potassium iodide (SSKI) for one day prior to and
for 6 days following administration of the tracer [13]. The
critical organ is the adrenal medulla (100 rad/
mCi 131I).
With the use of 123I-mIBG manufactured with low
free iodine (1-3%) and considering the 13.2 hour half-life of 123I,
extended thyroid blockade is probably unnecessary [37]. For 123I-mIBG
imaging, a single dose of SSKI (or the equivalent) is effective
in reducing the thyroid dose by more than 50% (best given 1 hour
prior to tracer administration) [37]. However, even with
blockade, some thyroid uptake does occur due to symathetic nerve
terminals in the thyroid gland [37]. The use of thyroid blockade
for 123I-mIBG can be individualized and may not be
required in elderly patients as the radiation burden to the
thyroid gland from the administration of 10 mCi of high-purity 123I-mIBG
has been estimated to be 70 mGy in nonblocked patients (siilar
to the dose received from an I-123 thyroid scan) [37]. Prior to
I-123 MIBG imaging, thyroid blockade can be performed with a
saturated solution of potassium iodide 100mg/day starting 1 day
before tracer injection and continuing for 2 days [39].
For I-123 imaging, planar continuous whole body images are acquired using a scan speed of 6.77 cm/min and SPECT images can be obtained if indicated [24]. For I-131 MIBG imaging, planar static images are obtained over the chest, abdomen, and pelvis for 20 minutes each using a dual headed camera, a high energy collimator, and a 20% window centered at the 364 keV photopeak [24]. Spot images are prefered to a continuous acquisition due to the low count rate [24].
Drugs
which interfere with MIBG uptake
- Cocaine: Inhibits
catecholamine uptake [12]
- Tricyclic
antidepressants (Desipramine, Amitriptyline [Elavil]):
Inhibit
catecholamine
uptake
[12]
and should be discontinued for 3-4 days prior to the
procedure [24].
- Phenylpropanolamine/Pseudephredine/Phenylephrine-
found in over the counter cold preparations and
decongestants these agents deplete storage vesicle contents
- Catecholamine
agonists: Sympathomimetics,
Amphetamines, and Amphetamine-like compounds. Deplete
storage vesicle contents [12]
- Reserpine: Depletes
the catecholamine stores in the neurosecretory
storage granules and inhibits vesicle active transport [12].
It should be discontinued for 3-4 days prior to the
procedure [24].
- Antipsychotics: Phenothiazines (Thorazine)
and thiothixines inhibit
catecholamine uptake [12]
- Calcium channel
blockers: Should be discontinued for 3-4 days prior to the
procedure [24].
- Adrenergic
blockers: Long acting beta-blockers: Labetalol
and possibly metoprolol. Labetalol inhibits catecholamine
uptake and depletes storage vesicle contents [12]. It should
be discontinued for at least 3 weeks prior to scanning. The
absence of salivary and parotid activity can serve as a clue
that patients may be on this medication.
- Possibly some
foods: Those which contain vanillin and catecholamine-like
compounds such as chocolate and blue-veined cheeses
Disease Processes and Neoplasms
Imaged with MIBG:
Pheochromocytoma
Clinical
Presentation
Pheochromocytomas are a rare tumor
(incidence of 0.05% in the general population in an autopsy
series [36]) that arise from chromaffin
cells (neural crest ectoderm involved with catecholamine
synthesis) which is found in the adrenal medulla (85-90% of
tumors arise in the adrenal medulla), sympathetic ganglia, Organ
of Z?ckerkandl, and the aortic and
carotid chemoreceptors. Patients
with sporadic pheos are older at presentation with a mean age of
44 years [36]. Patients can present with paroxysmal HTN (50%) or
sustained HTN (40-90%) associated with headache, sweating,
palpitations, weight loss, and hyperglycemia (Note: Only 0.1% to
0.6% of patients with hypertension are found to have pheochromocytomas [18,36]). Symptoms may
be precipitated by abdominal exertion such as heavy lifting or
performing the Valsalva maneuver
[17]. The clinical triad of headache, palpatations, and sweating
are 90% specific for pheochromocytoma, the triad is uncommon-
occurring in 10-36.5% of patients [36]. Other symptoms include
nausea, flushing, weight loss, fatigue, and hyperglycemia [36].
Men and women are affected equally [18]. Pheos are exceptionally
rare in the pediatric population, however, pediatric pheos have
a higher incidence of malignancy (47% versus 10% in adults)
[36]. Glucagon is contraindicated in these
patients as they may cause catecholamine release from the tumor
and precipitate a hypertensive crisis. Biopsy is also
generally not indicated for patients suspected of having pheochromocytoma- due to the risk of
precipitating a hypertensive crisis [26]. The prognosis is very
favorable for patients with benign lesions [36]. Classic
indicators of poor prognosis include tumor size greater than 5
cm, extraadrenal tumor location, age younger than 30 years at
first presentation, and metastatic disease [40]. The prognosis
is poor for patients with malignant pheochromocytoma with a 5
year survival rate of 54% [36].
Laboratory abnormalities include
elevated urinary and resting plasma catecholamine levels (97%
and 99% sensitivity, respectively [21]), and elevated 24-hour
urine vanillylmandelic acid (VMA)
(50-90%) and metanepinephrine
levels. These tests have sensitivities ranging from 89-100%
[15]. Serum chromogranin A (CgA- a chromogranin
protein which is cosecreted with
neurotransmitters) is also an effective marker for pheochromocytoma [14].
Pheochromocytomas follow a rule of
"ten": 10% are malignant (tend to be larger lesions), 10% are
bilateral, 10% occur in pediatric patients, and 10% are
extra-adrenal. However, due to advances in
molecular diagnostics, it is likely that about 25% of
pheochromocytomas have a genetic association [36]. Clinical
features that may be associated with an increased risk for a
malignant tumor include high preoperative 24 hour urinary
dopamine levels, extraadrenal location, tumor weight > 80
gm, and persistent post operative hypertension [36].
Pheochromocytomas are associated
with certain genetic disorders and affected patients typically
present at a younger age (mean 25 years) [36]. Associated
disorders include: Neurofibromatosis Type I (develop in
approximately 1% of patients [36]), von Hippel-Lindau
(develop in 10-26% of patients [36]), MEN syndromes IIa (50% prevalence [21]) & MEN IIb (90% prevalence [21]), and Carney's
syndrome (which consists of smooth muscle tumors of the stomach,
chondromatous pulmonary hamartomas, and extra-adrenal paragangliomas) [17]. Pheochromocytomas associated with MEN
syndrome tend to be bilateral (bilateral
pheochromocytomas are found in up to
75% of these cases) and are almost always intraadrenal-
they also tend to be benign [18]. Pheochromocytoma-paraganglioma
syndromes are also associated with succinate dehydrogenase
mutations (SDHB and SDHD) [36]. These gene mutations are found
in about 10% of patients with pheochromocytoma [36]. Patients
with SDHB mutation are more likely to have extra-adrenal tumors
and head and neck paragangliomas [36]. Additonally, patients
with SDHB mutations have up to a 50% risk for malignant pheo
[36].
Extra-adrenal lesions occur in 10% of adults, and 30% of children. Extra-adrenal lesions are generally located in the retroperitoneal sympathetic ganglia in the paracaval/aortic region or near the organ of Z?ckerkandl. Other sites include the renal hilum (may have associated ipsilateral renal atrophy), the mediastinum (1%), and the urinary bladder (1%- associated with micturitional syncope). In adults, extra-adrenal lesions are malignant in 30-40% cases, while despite their higher prevalence, only about 2% of extra-adrenal lesions are malignant in children. The sensitivity of 131I-MIBG imaging is significantly lower in the diagnosis of exrtra-adrenal pheochromocytoma than in the diagnosis of adrenal pheochromocytoma [27].
Paragangliomatosis refers to the presence of "too numerous to count" pheochromocytomas. This condition is noted in about 5% of patients with extra-adrenal lesions. In this condition, the majority of the lesions are benign, but they may still be functionally active.
Radiographic
Findings
CT: CT can be performed safely with low osmolar, non-ionic I.V. contrast [18,23,36]. On CT most pheochromocytomas
are greater than 3 cm in size. They are usually soft tissue
density and the center of the lesion may be necrotic.
Approximately two-thrids of the lesions are solid and the rest
are complex or cystic [36]. The lesion may appear completely
cystic. Calcification is noted infrequently- although, other
authors indicate that calcificaiton can be seen in 10% of tumors
and up to 21% of symptomatic lesions [36]. The tumor is hypervascular and will exhibit marked
enhancement of the solid components [36].
MR: On MRI, pheochromocytomas have
a signal intensity similar to or
slightly lower than the liver on T1 images, but are extremely hyperintense on T2 images (up to 70% of
lesions show very bright T2 signal [26]). This classic
appearance is variable in prevalence- ranging from 11-65% of
lesions [36]. Occasionally, lesions may be inhomogeneous, iso-, or hypointense
to the liver. Up to 1/3 of lesions may have low signal on T2
images [36]. Unfortunately, due to the presence of necrosis
within the lesion, over 20% of lesions will have an atypical
appearance on T2 images [5]. On rare occasions, pheos may
contain microscopic fat resulting in signal loss on chemical
shift imaging (mimicking an adrenal adenoma) [36].
Angiography is contraindicated unless absolutely necessary as it may provoke a hypertensive crisis. In such cases, premedication is required. The lesion is typically hypervascular.
Scintigraphic Findings
In patients with biochemical abnormalities indicative of a pheochromocytoma, CT or MRI are more
accurate in detecting primary tumors of the adrenal gland, but
they are inferior to MIBG in the detection of extra-adrenal
tumors which account for about 10% of lesions [6]. I-131 MIBG
scan sensitivity for pheochromocytoma
is 56-90% and specificity is 95-100% [5,15,17,34].
The
sensitivity
of
I-123
MIBG
(sensitivity 76-100%; specificity 84-100% [25,29,39])
has
been reported to be higher than I-131 due to its superior
imaging characteristics [24]. It has been reported that the exam
sensitivity is lower for SDHB and VHL-related pheochromocytomas
and paragangliomas [39].
The sensitivity of 123I-MIBG is higher for the
identification of adrenal lesions than for extraadrenal
paragangliomas [29]. The sensitivity
of I123-MIBG for paragangliomas is between 56-76% and the
specificity between 84-100% [39].
I-123 MIBG also offers the option of performing SPECT imaging
to aid in lesion loicalization and
improved contrast resolution [25]. However, in one study, SPECT
imaging only produced a slight improvement in exam sensitivity,
while causing a slight drop in specificity [29]. The normal
adrenal medulla is visualized with I131 MIBG in about 10% of
cases, and in 50-80% of cases with I123 MIBG- and this
physiologic uptake may obscure small lesions [34]. Pathologic
adrenal uptake on MIBG imaging is usually more intense than the
liver or non-homogeneous [39]. MIBG imaging is more sensitive
for the detection of adrenal pheochromocytoma compared to
Octreotide imaging (while MIBG imaging has a lower sensitivity
for extra-adrenal paragangliomas) [34].
I-123 MIBG has been suggested to be less sensitivity for the evaluation of metastatic pheo when compared to Octreotide or 6-18F-Fluorodopamine PET imaging [25]. Approximately 10% of pheochromocytomas are cystic, but these lesions are still endocrinologically active and will concentrate MIBG. Unlike neuroblastoma, bone scan is superior to MIBG for detecting the presence of bone metastases in patients with pheochromocytoma.
Click here for discussion of PET imaging in pheochromocytoma
|
Pheochromocytoma: The patient was discovered to have a right adrenal mass (blue arrows) during a CT evaluation for ovarian carcinoma. Further clinical and laboratory evaluation revealed the patient to be hypertensive with elevated urinary catecholamines. The MIBG exam demonstrated intense tracer accumulation in this lesion (black arrows) which was proven to be a pheochromocytoma. No other lesions were identified. Thyroid activity can be seen due to inadequate thyroid blocking with SSKI drops. |
|
|


|
Pheochromocytoma: The patient below was being evaluated for hypertension and abnormally elevated catecholamine levels. CT revealed a mass in the left adrenal gland (white arrows). The 72 hour posterior MIBG image demonstrated abnormal tracer accumulation in this lesion (black arrows) consistent with a pheochromocytoma. Physiologic hepatic activity can be seen in the right upper abdomen. |
|
|


68Ga-DOTATE
68Ga-DOTATE can accumulate in pheochomocytomas and
paragangliomas [45]. Lesion detection appears to be very high in
patients with SDHB-related metastatic lesions [45].
Neuroblastoma
Clinical
Presentation
Neuroblastoma arises from neural
crest derivatives in the adrenal medulla or the sympathetic
nervous system [20]. It is the most common extracranial
solid malignant tumor of infants and children [19] and the 3rd
most common childhood malignancy overall (after leukemia and
primary CNS brain tumors [31]). It accounts for 10-15% of
childhood malignancies [31] and for nearly 15% of all pediatric
cancer fatalities [44]. About 50% of cases occur in children
less than 2 years of age [20] (the median
age at diagnosis is 19 months [44]), and 90% are seen
by ages 5-8 years. Males are affected slightly more than
females. Although there is no racial variation in incidence,
African-Americans are more likely to have high-risk disease and
fatal outcomes [41,44]. The majority of neuroblastomas are
sporadic, but about 1% are familial and are associated with
mutatuions in the ALK, PHOX2B, and KIF1B genes [44].
Neuroblastoma is also the most common malignancy in the first month of life and fetal/cogenital neuroblastoma (almost always adrenal in origin- 90% of cases) can also occur [16,44]. A unique perinatal variant is called cystic neuroblastoma which is characterized by one or more macroscopic cysts in the tumor that are felt to be related to prior hemorrhage/necrosis [43]. Associations with a familial predisposition (0.2%), bowel agangliosis, and congenital heart disease have been described, but none are very strong.
Symptoms of neuroblastoma include
an abdominal mass, limp, nystagmus,
or inability to walk. Hypertension is found in 30% of patients.
Elevated urinary catecholamine levels (VMA & HVA) are seen
in 75-90% of patients.
Metastases are present in 60-70% of patients at the time of
diagnosis [19,31]. The skeleton or
bone marrow is the most common site of metastatic disease (60%) (70% bone marrow and 55% bone [33])-
when associated with irritability and limping this is known as
Hutchinson syndrome [16]. Metastatic involvement of the skull
can occur in up to 25% of cases [31]. Metastases can also go to
lymph nodes (35-40% at the time of diagnosis), the liver, and
intracranial (parenchymal or leptomeningeal metastases are found in
2-16% of cases [31]). Skin metastases are relatively common,
especially in children younger than 1 year of age [16]. Skin
lesions may appear as dark blue or purple masses- resembling
blueberries- this appearance has been given the eponym
"blueberry muffin syndrome" [16]. Ecchymotic
orbital proptosis ("raccoon eyes")
results from metastases to the perioribital
bones and soft tissues causing obstruction of ophthalmic and
facial veins [19,44].
Paraneoplastic syndromes are also seen and include:
- Infantile myoclonic encephalopathy (opsoclonus myoclonus ataxia syndrome): The condition occurs in 2-3% of neuroblastoma patients [44], but it is associated with occult neuroblastoma in 20-50% of cases. The syndrome consists of opsoclonus (random irregular eye movements or "dancing eyes"), myoclonus (rhythmic jerking movements of the extremitis), and cerebellar ataxia. The condition is generally associated with favorable well-differentiated neuroblastomas [41] and is believe to be immune mediated through antibodies that react with central nervous system antigens [44]. Tumor removal does not always improve neurologic symptoms, but immunotherapy with IV immunoglobulin can improve neurologic symptoms in up to 83% of patients during intial treatment, however, relapse is common [44].
- Verner-Morrison syndrome: Watery diarrhea, hypokalemia, and achlorhydria. Seen in 5% of cases, it is secondary to VIP (vasoactive intestinal peptide) production by the tumor (elevated VIP levels are noted in 25% of patients with neuroblastoma). The symptoms will improve following tumor resection [44]
Sites of Primary Neuroblastoma: 65% to 75% of cases are intraabdominal [41].
Approximately 80% of tumors occur in the abdomen [48]. Primary
tumor location is associated with outcome with a higher risk for
relapse with adrenal tumors and a lower risk for thoracic tumors
[41].
Adrenals: Most frequent (35% to 66% of cases) intra-abdominal site. About 10% of cases are bilateral. Primary adrenal lesions are actually associated with a worse prognosis than extra-adrenal lesions.
Extra-adrenal abdominal sympathetic ganglia: Includes the Organ of Z?ckerkandl (a cluster of paraganglia near the aortic bifurcation, seen predominantly in children).
Intrathoracic: The mediastinum is the second most common location for primary neuroblastoma after the abdomen, most commonly the posterior mediastinum [42]. Approximately 15% to 20% of cases are intrathoracic and intrathoracic tumors are more common in infants [41]. The lesion generally arises posteriorly in the paravertebral sympathetic chain. Between 5-25% of patients will have an associated intraspinal component. Thoracic and cervical primaries are associated with a better prognosis.
Neck: Accounts for 1% to 5% of cases
Intracranial: Rare (approximately 2% of cases). Most commonly arises in the olfactory nerve (esthesioneuroblastoma), or cerebellum.
Staging [19], Treatment, and Prognosis of Neuroblastoma
INSS staging
- Stage I: Confined
to the organ of origin with complete gross surgical excision
- Stage IIA:
Localized tumor with incomplete gross excision
- Stage IIB:
Localized tumor with complete of incomplete gross excision.
Tumor extends into adjacent structures, but does not cross
midline. Ipsilateral regional
lymph nodes positive for tumor, but contralateral
nodes are negative.
- Stage III: Unresectable tumor which crosses
midline; or unilateral tumor with contralateral
regional lymph node involvement
- Stage IV: Distant
metastases
- Stage IVS: Localized primary lesion that does not cross midline, with metastases to liver, skin, and/or bone marrow (but NOT cortical bone) in infants younger than 1 year of age. Stage IVS disease is associated with very good survival following chemotherapy and radiation.
INRGSS staging [41]
L1- Localized tumor not involving vital structures and confined to one body compartment
L2- Local-regional tumor with one or more image-defined risk factors
M- Distant metastatic dissemination
MS- Metastatic dissemination limited to skin, liver, and/or bone marrow in patients younger than 18 months
Prognosis: The most important predictors of prognosis are age
at presentation and stage [16]. Children
who are less than one year of age have a significantly higher
5 year survival rate compared to patients diagnosed after the
age of one [44]. Similarly, a lower stage or stage IVS disease are associated with a
better prognosis [19]. Children with stage 1, 2, and 4S disease
have a 3 year event-free survival of 75-90% [16]. Children under
the age of 1 year with stage 3 and 4 tumors have a one-year
event free survival rate of 80-90% and 60-75% respectively [16].
Children older than 1 year with stage 3 and 4 tumors have a 3
year event-free survival rate of 50% and 15%, respectively [16].
Unfavorable prognostic features include age ≥ 18 months, the
presence of distant metastatic disease in patients older than
12-18 months (lymph nodes, bone or bone marrow, and liver), MYCN
oncogene amplification on molecular
pathology in patients of any age, and unfavorable
histopathologic features (high-risk neuroblastoma has a
long term survival rate of about 40%) [32,47]. A complete
response to induction chemotherapy is one of the main early
prognostic factors in patients with high risk neuroblastoma
[47]. A Curie score of greater than or equal to 2 at the end of
induction therapy is associated with an inferior outcome [44].
Following induction, the event free survival has been reported
to be 15% for anything more than minimal residual disease,
compared to 46% for patients with a Curie score of less than 2
[44]. Another semiquantitative scoring system (SIOPEN) with a
post induction skeletal score greater than 3 also identifies
patients with a very poor prognosis who require different
treatment strategies [47].
Tumor genetics: MYCN gene amplification occurs in about 20% of
primary tumors and is associated with advanced tumor stage,
rapid tumor progression, and poor outcome [44]. Chromosomal
deletions ar found in up to 50% of neuroblastomas and those
associated with a worse prognosis include deletion of 1p36,
unbalanced gain of 17q, and deletion of 11q [44]. Neuroblstomas
with a higher DNA content (DNA index > 1, hyperdiploid) are
associated with a lower tumor stage and improved outcomes in
children younger than 18 months [44].
Occasionally, the lesions may revert to a benign ganglioneuroma (0.2% to 1% of cases). Regression most often occurs in stage IVS and takes 6 to 12 months [16]. A persistently positive MIBG scan during and after induction therapy is associated with a poor outcome [19].
Therapy:Surgery is the mainstay of treatment for low-risk tumors [44]. Chemotherapy is added for tumors that cannot be resected, as well as for intermediate risk patients [44]. For high risk patients, an aggressive multimodality treatment regimen is used combining chemotherapy, surgical resection, adjuvant high-dose chemotherapy with hematopoietic stem cell rescue, and radiation therapy [44]. Maintenance therapy entails the use of anti-GD2-immunotherapy with dinutuximab [44].
I-131 MIBG has been used in therapeutic doses as part of a multimodality approach for the treatment of neuroblastoma- predominatly in refractory and relapsed patients [30,41]. Up to 14% of patients receiving conventional I-131 MIBG therapy can have severe acute hypertension during the infusion likely due to unlabeled MIBG [46]. The outcome of the treatment has been variable with reported partial responses raning from 15-92% [30,46]. Myelosuppression is the main toxic effect and autologous peripheral blood stem cell rescue is often necessary after higher administered doses [41]. Thyroid disorders (hypothyroidism and nodules) are common after I131-MIBG therapy and continuous thyroid screening should be performed in these patients [41].
Scintigraphy in the Evaluation of Neuroblastoma
Agents: Either I-131 MIBG or I-123 MIBG can be used for imaging [20]. I-123 MIBG has a more favorable dosimetry (whole body radiation dose in about 5% of that of I-131 MIBG) and better imaging characteristics- including higher quality images, greater sensitivity for disease detection, lower risk to the thyroid gland, and the ability to perform SPECT imaging [20,28]. As a result, I-123 MIBG has essentially replaced I-131 MIBG for imaging.
Patient preparation/imaging: A solution of potassium
iodide is administered to reduce thyroid uptake of free
radioiodine, beginning the day before injection and continued
for 5 to 6 days. The recommended doses are 32 mg potassium
iodide daily for children aged 1 month to 3 years, 65 mg for
those 3 to 13 years, and 130 mg for those older than 13 years
[20]. The additional use of a single dose of thyroxine (100 mcg/m2) and
two doses of methimazole
(0.25mg/kg/m2) can also be administered [20].
I-131MIBG (0.5-1.0 mCi/1.7 m2
body surface area [7 to 15 uCi/kg]) is administered by slow IV
injection over 90 seconds. Spot images of the entire body
(chest, abdomen, and pelvis) should be obtained at 24-48 hours
following tracer administration [20]. Images are acquired for
100,000 counts or 20 minutes, whichever comes first. A
low-speed, whole body gamma camera can also be used to obtain
the images. [12]
For I-123 MIBG, a saturated solution of potassium iodide
should be given the day before administration of the tracer and
continued for 1 to 3 days [41]. A single dose of saturated
solution of potassium iodide at the time of tracer injection has
also been shown to adequately block thyroid uptake [41]. The
dose of I-123 MIBG is 0.14 mCi/kg (5.2 MBq/kg) with a minimum
administered activity of 1 mCi (370 MBq) [41]. I-123 MIBG
imaging is performed 24 hours after tracer administration and at
48 hours if needed [41].
Image interpretation:
The modified Curie score is used for semiquantitative image
interpretation [44]. The body is divided into nine skeletal
segments with an additional segment for soft-tissue invovlement
[44]. Each segment is assigned a score of 0-3 depending on the
extent of disease involvement [44]. No involvement receives a
score of 0; single site disease in the segment a score of 1; two
or more sites of disease involving less than 50% of a segment
receives a score of 2; and more than 50% involvement of a
segment is given a score of 3 [44]. The maximum score is 30 and
the score on post therapy imaging has prognostic significance
[44].
Findings:
The overall sensitivity of MIBG in the detection of neuroblastoma is approximately 90%
(about 10% of neuroblastomas do not show MIBG uptake, although
other authors suggest that up to 30% may not show MIBG uptake
[42]) [7,12,38,41]. The overall
specificity of MIBG in the detection of neuroblastoma
is 94% [12]. The extent of metastatic bone and bone marrow
disease is most accurately defined by MIBG scintigraphy
(sensitivity 90%, specificity 100%) [33]. Bone scanning
often grossly underestimates the extent of osseous metastases
and MIBG is superior to MDP for detecting the presence of
skeletal metastases (likely because of the propensity for neuroblastoma metastases to originate
within the bone marrow cavity and high physiologic physeal
activity in children that can mask metastatic lesions) [8,12,33]. Bone scan has a reported
sensitivity of 70-78% and a specificity of 51% for the detection
of bone metastases [33].
Normalization of MIBG exams in patients with documented bone
marrow metastases is a better predictor of successful treatment
than MRI exams which can demonstrate persistent abnormal marrow
signal despite the absence of metastases for up to 2 years [9].
Liver metastases may escape detection with MIBG imaging. This
finding is not surprising as MIBG is physiologically
concentrated in the liver, and therefore small lesions may be
obscured [10]. SPECT increases diagnostic certainty, but does
not increase lesion detection with I123-MIBG [11]. Recurrent neuroblastoma may not demonstrate MIBG
accumulation in up to 50% of patients, suggesting that the role
of MIBG may be more limited in this setting [16].
False positive MIBG exams have been described in association
with atelectasis, pneumonia, focal nodular hyperplasia in the
liver, radiation induced liver injury, accessory spleen, focal
pyelonephritis, and vascular malformations [41].
Other agents:
TcMDP is concentrated in about two-thirds of primary lesions (and is felt to be related to presence of calcification). Bone scan detects the presence of skeletal metastases in 40-60% of patients, but localization of metaphyseal metastases (a common site of metastatic disease) can be difficult due to the adjacent physeal activity. A common finding on bone scan in patients with metaphyseal metastases is ill-defined increased activity in this area with loss of the sharp boundary between the growth plate and the metaphysis.
In-111 Octreotide can also be used to image neuroblastoma as MIBG negative paragangliomas will sometimes concenrtrate In-111 pentreotide [24]. Overall the agent has inferior sensitivity (55-70% [20]) compared to MIBG [19], and specificity also suffers as non-neuroendocrine tumors such as lymphoma and inflammatory infiltrates may also demonstrate tracer uptake [12].
PET:
FDG imaging:
Neuroblastomas are metabolically
active tumors [22] that demonstrate moderate FDG uptake (higher
stage lesions tend to show higher uptake than early stage
disease [38]). PET scan findings correlate well with disease
extent as determined by MIBG scans and the scans can be
performed 60 minutes following tracer administration [19,22]. FDG PET has been found to be most
beneficial in tumors that fail to accumulate or only weakly
accumulate MIBG [28]. PET may also be better at identifying
small lesions and localizing anatomic sites of disease due to
its superior spatial resolution [19]. PET appears to be less
useful in the detection of stage IV neuroblastoma
because of suboptimal detection of marrow disease compared to
MIBG scintigraphy due to normal FDG
activity in hematopoietic bone marrow [22,28,32,38].
In one study, the sensitivity of FDG was 86% [32].
18F-FDOPA PET imaging in
neuroblastoma:
FDOPA resembles natural L-DOPA [35]. The short-term
accumulation of FDOPA is determined mainly by the reaction rate
of amino acid decacrboxylase (AADC) which converts FDOPA into
fluorodopamine, which is then stored in vesicles [35]. FDOPA
imaging is very sensitive for the detection of neuroblasic
tumors including neuroblastomas, ganglioneuromas, and
ganglioneuromas [35]. Higher FDOPA upatake is associated with
higher levels of AADC expression and urinary VMA levels [35].
Neuroblastic tumors have AADC activity and can be imaged by
FDOPA PET [35]. Pre-treatment with carbidopa, an AADC inhibitor,
one hour before injection of FDOPA improves availability of
FDOPA for imaging [35]. FDOPA imaging can be performed 90
minutes following tracer administration, as opposed to I-123
imaging which requires a 24 hour period after injection prior to
imaging [35].
Normal FDOPA uptake can be seen in the basal ganglia,
gallbladder, kidneys, urinary tract, liver, and epiphyseal
plates of the long bones [35]. Low level GI tract activity can
also be seen [35]. Genitourinary and gallbladder activity are
seen due to excretion of catecholamine metabolites [35].
FDOPA is the most sensitive and accurate method of identifying
neuroblastoma localizations [47]. FDOPA has been shown to be
more sensitive than 123I-MIBG in staging
neuroblastoma patients and in evaluating disease persistence
after chemotherapy [47].
68Ga-DOTATE:
68Ga-DOTATE can accumulate in neuroblastoma lesions
[45]. SSTR expression (specificically SSTR type 2) can be seen
in 77-89% of neuroblastomas [45].
124I-MIBG:
124I is a positron emitting radionuclide with a
half-life of 4.2 days (compared to 13.2 hours for 123I)
[48]. This results in higher patient radiation exposure with an
effective dose twice that of an 123I-MIBG scan when
a very low activity is used for the exam [48] (the effective
dose has been calculated to be 0.161-0.795 mSv/MBq which is
approximately 10-20 times the effective dose of 18F-FDG
in the pediatric population [48]).
124I-MIBG has been shown to have superior lesion detection compared to 123I-MIBG imaging [48]. Increased lesion detection using 124I-MIBG has been correlated with higher Curie scores [48].
Correlative Radiographic Imaging in Neuroblastoma
Plain film findings in neuroblastoma include multicentric lytic skeletal lesions, metaphyseal lucent bands, and vertebral body collapse. Calcification is evident in at least 30% of cases [16]. On IVP there is typically inferior renal displacement rather than collecting system distortion (seen in Wilm's)
On CT neuroblastoma typically appears as a large complex mass which is often locally invasive into adjacent tissue and typically encases the great vessels, although a pseudocapsule maybe seen. Neuroblastomas often contain areas of necrosis, hemorrhage, and calcification (seen in between 50% to 80-85%). Calcification is usually coarse, amorphous, and mottled, but can be curvilinear or nodular. The mass may appear cystic and can mimic an adrenal hemorrhage in neonates. Neuroblastomas appear bright on T2-weighted MRI images.
|
Neuroblastoma: Bone scan (left) demonstrated clearly abnormal tracer accumulation within the right 5th rib. There is ill-defined tracer accumulation within the left upper abdomen. In retrospect, there is asymmetric tracer activity in the right iliac wing, the left femoral neck and the distal left femoral metaphysis. These lesions are more clearly demonstrated on the MIBG exam (center). A right distal femoral metaphyseal lesion is also identified. Markedly abnormal MIBG accumulation can be seen in the patients large neuroblastoma (CT scan right). |
|
|



|
Neuroblastoma: Bone scan and MIBG exam. Diffuse skeletal metastases are evident on bone scan, but are more clearly revealed on the MIBG exam. Uptake within the patients primary lesion (CT center white arrows) can also be seen on the MIBG exam. A plain film skeletal survey demonstrated a very subtle periosteal reaction within the distal left femoral metaphysis, but was otherwise normal (Click plain film to enlarge) |
|
|



Adrenal Medullary
Hyperplasia
In patients with adrenal medullary
hyperplasia, the level of accumulation of the radiotracer is
related to the degree of medullary
dysfunction. The exam typically reveals symmetric or nearly
symmetric increased tracer uptake within the glands, with
relative preservation of the glandular shape. Since the right
adrenal gland is more superficial on posterior images, it may
appear more intense than the left side.
REFERENCES:
(1) J Nucl Med 1998; Roelants V, et al. Iodine-131-MIBG scintigraphy in adults: Interpretation revisited? 39: 1007-1012
(2) J Nucl Med 1994; Mozley PD, et al. The
efficacy of iodine-123-MIBG as a screening test for pheochromocytoma.
35: 1138-1144
(3) Clin Nucl Med 1995; Elgazzar AH, et al. I-123 MIBG scintigraphy in adults. A report of clinical experience. 20(2): 147-152
(4) J Nucl Med 1994; Bonnin F, et al. Refining interpretation of MIBG scans in children. 35(5):803-10
(5) Semin Nucl Med 1995; Freitas JE. Adrenal cortical and medullary imaging. 25(3):235-50
(6) J Nucl Med 1993; Maurea S, et al. Iodine-131-metaiodobenzylguanidine scintigraphy in preoperative and postoperative evaluation of paragangliomas: comparison with CT and MRI. 34: 173-79
(7) Semin Nucl Med 1993; Gelfand MJ. Meta-iodobenzylguanidine in children. 23(3):231-42
(8) J Nucl Med 1993; Parisi MT, et al. Optimized diagnostic strategy for neuroblastoma in opsoclonus-myoclonus. 34(11):1922-6
(9) J Nucl Med 1997; Lebtahi N, et al. Evaluating bone marrow metastasis of neuroblastoma with Iodine-123-MIBG scintigraphy and MRI. 38: 1389-1392
(10) J Nucl Med 1994; Maurea S, et al. Iodine-131-MIBG imaging to monitor chemotherapy response in advanced neuroblastoma: comparison with laboratory analysis.35(9):1429-35.
(11) J Nucl Med 1994; Gelfand MJ, et al. Iodine-123-MIBG SPECT versus planar imaging in children with neural crest tumors. 35(11):1753-7
(12) J Nucl Med 1998; Shulkin BL, et al. Current concepts on the diagnostic use of MIBG in children. 39: 679-688
(13) Applied Radiology 2001; Hanson MW. Scintigraphic evaluation of neuroendocrine tumors. 30: 11-17
(14) J Nucl Med 2001; d'Herbomez M, et al. Chromogranin A assay and 131I-MIBG scintigraphy for diagnosis and follow-up of pheochromocytoma. 42: 993-997
(15) RadioGraphics 2001; Mayo-Smith WW, et al. From the RSNA refresher courses. State-of-the-art adrenal imaging. 21: 995-1012
(16) Radiographics 2002; Lonergan GF, et al. From the archives of the AFIP. Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: radiologic-pathologic correlation. 22: 911-934
(17) Radiographics 2002; Lee TH, et al. Best cases from the AFIP. Cystic pheochromocytoma. 22: 935-940
(18) Radiographics 2003; Rha SE, et al. Neurogenic tumors in the abdomen: tumor types and imaging characteristics. 23: 29-43
(19) J Nucl Med 2004; Kushner BH. Neuroblastoma: a disease requiring a multitude of imaging studies. 45: 1172-1188
(20) J Nucl Med 2005; Pashankar FD, et al. MIBG and somatostatin receptor analogs in children: current concepts on diagnostic and therapeutic use. 46: 55S-61S
(21) AJR 2005; Elsayes KM, et al. MRI of adrenal and extradrenal pheochromocytoma. 184: 860-867
(22) Radiol Clin N Am 2005; Jadvar H, et al. PET in pediatric diseases. 43: 135-152
(23) Radiographics 2006; Scarsbrook AF, et al. Multiple endocrine neoplasia: spectrum of radiologic appearances and discussion of a multitechnique imaging approach. 26: 433-451
(24) Radiographics 2007; Intenzo CM, et al. Scintigraphic imaging of body neuroendocrine tumors. 27: 1355-1369
(25) J Nucl Med 2008; Ilias I, et al. Comparison of 6-18F-Fluorodopamine PET with 123I-metaiodobenzylguanidine and 111In-pentreotide scintigraphy in localization of nonmetastatic and metastatic pheochromocytoma. 49: 1613-1619
(26) Radiology 2008; Boland GW, et al. Incidental adrenal lesions: princioles, techniques, and algorithms for imaging characterization. 249: 756-775
(27) J Nucl Med 1009; 99mTc-HYNIC-TOC scinitgraphy is superior to 131I-MIBG imaging in the evaluation of extraadrenal pheochromocytoma. 50: 397-400
(28) J Nucl Med 2009; Sharp SE, et al. 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. 50: 1237-1243
(29) J Nucl Med 2009; Wiseman GA, et al. Usefulness of 123I-MIBG scintigraphy in the evaluation of patients with kown or suspected primary or metastatic pheochromocytoma or paraganglioma: results from a prospective multicenter trial. 50: 1448-1454
(30) J Nucl Med 2009; Buckley SE, et al. Whole-body dosimetry for individualized treatment planning for 131I-MIBG radionuclide therapy for neuroblastoma. 50: 1518-1524
(31) AJR 2010; D'Ambrosio ND, et al. IMaging metastatic CNS neuroblastoma. 194: 1223-1229
(32) J Nucl Med 2011; Papathanasiou ND, et al. 18F-FDG
PET/CT and 123I-metaiodobenzylguanidine imaging in
high-risk neuroblastoma: diagnostic
comparison and survival analysis. 52: 519-525
(33) Radiology 2011; Brisse HJ, et al. Guidelines for imaging
and staging of neuroblastic tumors: consensus reort from the
International neuroblastoma risk group project. 261: 243-257
(34) J Nucl Med 2012; Taieb D, et al. Modern nuclear imaging
for paragangliomas: beyond SPECT. 53: 264-274
(35) J Nucl Med 2013; Lu MY, et al. Characterization of
neuroblastic tumors using 18F-FDOPA PET. 54: 42-49
(36) AJR 2013; Leung K, et al. Pheochromocytoma: the range of
appearances on ultrasound, CT, MRI, and functional imaging. 200:
370-378
(37) J Nucl Med 2014; Friedman NC, et al. Efficacy of thyroid
blockade on thyroid radioiodine uptake in 123I-mIBG
imaging. 55: 211-215
(38) J Nucl Med 2015; Uslu L, et al. Value of 18F-FDG
PPET and PET/CT for evaluaiton of pediatric malignancies. 56:
274-286
(39) J Nucl Med 2015; van Berkel A, et al. Semiquantitative 123I-metaiodobenzylguanidine
scintigraphy to distinguish pheochromocytoma and paraganglioma
from physiologic adrenal uptake and its correlation with
genotype-dependent expression of catecholamine transporters. 56:
839-846
(40) J Nucl Med 2015; Blanchet EM, et al. 18F-FLT
PET/CT in the evaluation of pheochromocytomas and
paragangliomas: a pilot study. 56: 1849-1854
(41) Radiographics 2016; Sharp SE, et al.
MIBG in neuroblastoma diagnostic imaging and therapy. 36:
258-278
(42) AJR 2016; Pavlus JD, et al.
Imaging of thoracic neurogenic tumors. 207: 552-561
(43) AJR 2016; Hwang Sm, et al. Congenital adrenal
neuroblastoma with and without cystic change: differentiating
features with an emphysis on the value of ultrasound. 207:
1105-1111
(44) Radiographics 2018; Swift CC, et al. Updates in diagnosis,
management, and treatment of neuroblastoma. 38: 566-580
(45) AJR 2018; Sanli Y, et al. Neuroendocrine tumor diagnosis
and management: 68Ga-DOTATE PET/CT. 211: 267-277
(46) J Nucl Med 2019; Pryma DA, et al. Efficacy and safety of
high-specific-activity 131I-MIBG therapy in patients
with advanced pheochromocytoma or para ganglioma. 60: 623-630
(47) J Nucl Med 2020; Piccardo A, et al. Diagnosis, treatment
response, and prognosis: the role of 18F-DOPA PET/CT
in children affected by neuroblastoma in comparison with 123I-mIBG
scan: the first prospective study. 61: 367-374
(48) J Nucl Med 2021; Aboian MS, et al. 124I-MIBG PET/CT to monitor metastatic disease in children with relapsed neuroblastoma. 62: 43-47