A breast imaging technology called breast transillumination will continue to be regulated under the U.S. Food and Drug Administration's (FDA) most stringent category for medical devices after an FDA panel declined a U.K. company's petition to relax its rules.
The FDA panel decided on April 12 in consensus that breast transilluminators should continue to be regulated as class III medical devices, which require premarket approval (PMA) applications with supporting data from clinical trials. Most medical imaging devices are considered class II devices, which usually are cleared via the 510(k) process.
In making the ruling, the panel decided that not enough was known about breast transilluminators to reclassify them as class II devices. More clinical studies should be performed to shed more light on the technology.
Low-intensity light
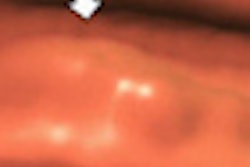
Transilluminators are electronic devices that use low-intensity emissions of visible light and near-infrared radiation (approximately 700 to 1,050 nm) transmitted through the breast to visualize translucent tissue for diagnosing cancer and other diseases or abnormalities, according to the FDA. When used properly, the transilluminator is intended to illuminate the vasculature of the breast; abnormalities such as a cyst or breast cancer would appear to the user as an area of darker absorption.
 |
| Breast transilluminators transmit low-intensity visible light and near-infrared radiation through the breast to visualize translucent tissue. All images are from FDA meeting materials submitted by PWB Health UK. |
In 1995, the FDA published a final rule that placed breast transilluminators in the class III category based on the recommendation of its Obstetrics and Gynecology Devices Panel, which concluded there were no published studies or clinical data demonstrating the safety and effectiveness of the devices.
Class III devices are defined as products that present "a potential, unreasonable risk of illness or injury," according to the FDA. Insufficient information exists for a class III device, such that performance standards (class II) or general controls (class I) cannot ensure that the device is safe and effective for its intended use. Full-field digital mammography units are currently categorized as class II.
Fifteen years after the initial classification, the FDA issued a proposed rule calling for PMA applications for breast transilluminators. In response, it received a petition from U.K.-based PWB Health, makers of Breastlight, a handheld device that transilluminates the breast with a red light of 617 nm; the light is absorbed by hemoglobin so that areas of high vascularity (such as malignant tumors) appear black, according to PWB.
 PWB Health has sold 28,000 Breastlight devices outside the U.S.
PWB Health has sold 28,000 Breastlight devices outside the U.S.
The company asked that breast transillumination devices be downgraded to class I, stating that the units are already categorized as class I devices in Canada and the European Union. The petition also stated that the risks of breast transilluminators that were of concern to the FDA's Obstetrics and Gynecology Devices Panel (electrical shock, optical radiation, and the potential for missed or delayed diagnosis) have been adequately addressed.
In its April 12 meeting, the FDA's Radiological Devices Panel of the Medical Devices Advisory Committee met to review the Breastlight petition, particularly the classification of breast illuminators and whether they should be reclassified. The committee discussion focused on a review of available literature to assess the current knowledge of breast transilluminators and to determine if sufficient safety and effectiveness data are available to support their reclassification.
To address the petition, the FDA's Division of Epidemiology, Office of Surveillance and Biometrics, conducted a literature review, analyzing studies on the technology published between January 1995 and February 2012. The review determined the following:
- Rates of false positives and false negatives for breast transilluminators in asymptomatic women with nonpalpable masses could not be determined.
- Current literature does not address safety concerns related to misdiagnosis with resulting delayed treatment or unnecessary biopsies.
- Additional clinical studies are needed to provide valid scientific evidence to establish the effectiveness of breast transilluminators according to their indication in the intended population.
PWB Health's own materials submitted for the panel hearing state that the company has sold 28,000 of the devices in the U.K., Europe, Middle East, Africa, and China. The handheld device is designed to be used in the home as a means of encouraging women to monitor their own breast health on a regular basis. It's particularly well-suited for women with "naturally lumpy" or fibrous breasts, or to check for interval cancers between x-ray mammography screening studies.
The company has conducted three postmarket surveillance studies in more than 1,400 users. One study of 300 patients found that 12 of 18 malignant tumors were detected using Breastlight, for a sensitivity of 67%, while 240 of 282 breasts with no malignancy were correctly identified as negative, for a specificity of 85%.
The PWB Health materials state that while Breastlight is "not a suitable alternative to x-ray mammography," it is a useful addition to breast awareness and self-examination. The company's first study of more than 1,000 women found Breastlight to contribute to an "increase in breast awareness/breast self-examination" in 33% of study participants.
Benefit-to-risk ratio
The panel discussed the risks associated with breast transilluminators to determine their appropriate classification, as well as the benefit-to-risk ratio and the devices' potential impact on public health, according to an agenda the FDA released.
In its recommendations, achieved by consensus, the panel stated that it generally believes that breast transillumination devices carry risk of missed diagnosis, delayed diagnosis, delayed treatment, electrical shock, and optical radiation -- and there may be other risks including anxiety, thermal injury, eye injury, and overdetection. The panel also stated that class I general controls and class II special controls are not sufficient for breast transillumination devices, and it confirmed that these devices should remain as class III.
"The key idea is that these are a broad class of devices, and we don't know much about them," Dr. Robert Rosenberg, committee chair, told AuntMinnie.com. "None of them are being used in the U.S., so there just isn't much research to go on."