Monoclonal Antibody Therapy:
Radioimmunotherapy uses monoclonal antibodies (mAb) directed against specific tumors antigens labeled with a particle emitting radioisotope to deliver radiation directly to the tumor [15].
The two most common radioisotopes used for labeling monoclonal antibodies are I-131 and Y-90. I-131 is low cost and because of its gamma emission, images can be obtained revealing the agents biodistribution [5]. Disadvantages of I-131 include radiation exposure to other individuals as a result of the gamma-emission and dehalogenation due to metabolism of the mAb (the release of free I-131 requires thyroid protection to prevent hypothyroidism) [5]. Y-90 is a pure beta emitter and hence, imaging cannot be performed with this isotope [5]. The benefit of using Y-90 is that there is negligible radiation exposure to other individuals- even with very high doses [5]. Y-90 beta-particles also have a longer path length compared to I-131 which is advantageous in tumors with heterogeneous antibody distribution (max path length Y-90 is 11 mm [avg 2.5 mm] compared to 2.9 mm for I-131 [avg 0.4 mm]) [5].
Although monoclonal antibody therapy of malignancies would appear to be the ideal treatment modality, there are a number of problems which complicate their use:
- Tumor type: Tumor response depends on numerous variables including penetration, tumor radiosensitivity, dose rate, and cumulative dose. Solid tumors have not been as responsive to monoclonal antibody therapy as hematopoietic neoplasms [2]. In general, tumor uptake of the agents is low (0.001% to 0.01% of the injected dose per gram of tumor) [2]. Therefore, a cumulative tumor dose is generally less than 1500 cGy and large volumes of antibodies are available for interaction with normal host tissue [2].
- Dose limiting myelotoxicity has been a complication identified in most clinical trials, despite a relatively low dose of radiation delivered to the tumor. The marrow dose limit is about 150-200 cGy [2].
- Limitations of I-131: The relatively short range of I-131 beta emissions (less than 2mm) compromise its ability to treat large solid tumors [13]. Additionally, there is dehalogenation of the agent from the antibody in vivo. Finally, because of it's gamma emission there are environmental safety concerns.
- Murine monoclonal antibodies elicit human antib-mouse antibody (HAMA) formation which can limit the effectiveness of re-treatment if necessary. HAMA formation usually does not cause significant adverse reactions, however, HAMA can significantly alter the murine antibodies pharmacokinetics through the formation of antibody/HAMA complexes. These complexes are quickly removed by the reticuloendothelial system which decreases circulating levels of antibody and impairs tumor targeting [5].
- Antibody fragments have improved tumor penetration compared to intact IgG and elicit less HAMA formation, but have shorter tumor residence times [2] and result in excessively high radiation doses to the kidneys.
- Chimeric antibodies represent "humanized" monoclonal antibodies that are considerably less immunogenic [5]. Chimeric antibodies are formed by cloning a variable region of a murine antibody into a human expression vector containing the appropriate human constant region [5]. Although human antichimeric antibody (HACA) formation can occur, it is much less frequent than HAMA formation [5].
Non-antibody tumor targeting:
Non-antibody agents have also been evaluated as possible therapeutic agents. I-131 MIBG has been evaluated in the treatment of pheochromocytomas and neuroblastoma. The half-life of MIBG in tumor deposits is long (3 to 8 days) relative to the average whole-body biological half-life. In the treatment of pheochromocytomas doses between 75 to 300 mCi have been used without significant hematologic toxicity. Following treatment, up to 60% of patients had improvement in symptoms, but a complete response occurred in only 3%. In the treatment of neuroblastoma, although initial response to the treatment is good, most remissions are partial and have been short lived. Large marrow doses occur in patients with tumor in this location and significant marrow depression and prolonged thrombocytopenia has resulted. Post therapy scans have demonstrate more foci of metastatic disease than can be appreciated on diagnostic scans.
The somatostatin analog Pentreotide (an 8 amino acid peptide) has also been evaluated for therapeutic use. Tumor uptake has been measured to be as high as 0.1 to 0.8% of the injected dose per gram of tumor [17].
Molecular tumor targeting using aptamers (small oligonucleotides that are selected to bind tightly and specifically to a target molecule) may offer an alternative to monoclonal antibody therapy [16]. Rapid tumor uptake and rapid blood clearance, as well as little uptake in non-target tissues, may make these agents potentially very useful for cancer therapy [16].
Lymphoma treatment:
The incidence of non-Hodgkin's lymphoma is increasing and affects between 50,000-60,000 patients per year [5]. Approximately 80% of lymphomas are of B-cell origin [5] and follicular lymphoma is the most common form of indolent non-Hodgkins lymphoma [25]. Most patients with low-grade non-Hodgkin's lymphoma present with advanced stage disease [11]. For asymptomatic patients with advanced stage disease, watchful waiting is the recommended strategy, whereas symptomatic patients can be treated with a single or multiagent chemotherapy [30]. Despite the fact that they present with advanced stage disease, most patients will achieve an initial response with chemotherapy [11,12]. Unfortunately, despite the high initial response rate, nearly all patients will relapse [5]. The response rate typically declines and the duration of response grows progressive shorter with each subsequent course of therapy (due to development of drug or drug combination resistance) [11,12,25]. Treatment options therefore become more limited in patients that have relapsed or refractory, low-grade, follicular, or transformed non-Hodgkin's lymphoma [8]. Additionally, between 33% to 60% of patients undergo a transformation into a higher grade tumor with an accelerated rate of growth and a poor prognosis [11,12].
Anti-lymphoma antibody therapy has been used in the treatment of
low grade non-Hodgkin's lymphoma. The unlabeled monoclonal
antibody rituximab mediates its therapeutic effects through
several mechanisms including anti-body dependent cellular
cytotoxicty, complement fixation, and the direct induction of
apotosis [32].
Radioimmunocongugates have been developed in an attempt to take advantage of targeted anti-lymphoma antibodies by providing additional local radiation effects [11]. Two agents presently available are Zevalin and Bexxar. Both agents target the CD20 antigen which is densely expressed on nearly all (more than 90% [18]) B-cell lymphomas, however, it is also expressed on normal mature B-cells [13]. CD20 is not expressed on hematopoietic stem cells, normal plasma cells, or other normal non-lymphatic tissues [13,24]. CD20 is also not rapidly internalized, and is not shed from the cell surface when antibody binds- features which make it an attractive target for radioimmunotherapy [13,24]. Myelotoxicity is a common complication of anti-lymphoma andtibody therapy [13]. A significant number of patients in the trials for Zevalin and Bexxar required supportive therapy, including platelet transfusions (22% and 15%, respecively) and erythropoietin or epoetin alfa (8% and 7%, respectively) [13].
FDG PET imaging can be used to monitor the effectiveness of radioimmunotherapy [23,26]. Patients with a greater than 52% drop in SUV max have a longer survival compared to patients with lesser declines (larger declines are associated with longer disease free survival) [23]. In patients who progress following radioimmunotherapy, it appears that new sites of disease commonly develop, as opposed to recurrence or progression at previous sites of disease [23]. See also below for treatment specific results.
Zevalin (90Y-ibritumomab tiuxetan):
Ibritumomab is an IgG1 kappa-monoclonal antibody directed against the CD20 antigen that is found on normal mature B-lymphocytes and on more than 90% of malignant B-lymphocytes in patients with B-cell non-Hodgkins lymphoma (NHL) [3,7]. The CD20 antigen is an optimal target for immunotherapy because it is absent from hematopoietic stem cells and antibody binding does not induce antigen shedding or modulation [6].
90Y-ibritumomab tiuxetan (Zevalin) is a radioimmunotherapeutic agent approved for treatment of relapsed or refractory low grade follicular, or CD20+ transformed NHL when marrow involvement is not more than 25% (as measured by bone biopsy) [1]. 90Y-ibritumomab tiuxetan consists of a murine monoclonal antibody covalently attached to a metal chelator (90Y) [1]. 90Y is a pure beta emitter that decays to 90Zr with a half life of 64 hours (2.7 days) [1]. It has a high beta energy (maximum energy 2.28 MeV and average energy 935 keV) and an effective path length of 5.3 mm- meaning 90% of its energy is absorbed within a sphere with a 5.3 mm radius (about 100-200 cell diameters) [1,7], but the particle range can be up to 12 mm [2]. Because of it's higher energy, the 90Y beta particle has a longer path length in tissue than does the 131I beta particle (average energy 183 keV and a path length of approximately 1 mm [32]) [23]. As a result, 90Y has a higher therapeutic efficiency ratio for tumors greater than 5 cm (based on Monte Carlo-based dosimetry studies) [23]. Unbound agent accumulates in bone so a stable antibody attachment is very important to limit myelotoxicity [2]. Materials with a low atomic number, such as plastic or acrylics, are ideal for shielding and the beta particles will be completely absorbed in approximately 1 cm of these materials [1].
Eligibility for treatment:
Zevalin is indicated for the treatment of patients with relapsed
or refractory low-grade, follicular, or CD20 (+) transformed
B-cell non-Hodgkins lymphoma [24]. Patients should not have had
prior myeloablative therapy and no prior radioimmunotherapy [6].
It has recently been shown that patients with advanced follicular
lymphoma who have responded to first-line therapy that receive
consolidation with Zevalin, have a high PR to CR conversion (77%
of patients in PR following induction converted to a CR) and a
significantly longer progression free survival (PFS was prolonged
by 2 years) [30,31].
Patients with more than 25% lymphoma marrow involvement (determined by bone marrow biopsy ideally performed within 6 weeks of the planned therapy [7]), platelet counts less than 100,000, absolute neutrophil counts less than 1500 cells/mm3, or impaired bone marrow reserve should NOT receive Zevalin therapy [3]. Patients with impaired marrow reserve include those who have been given prior myelotoxic therapies with autologous bone marrow transplantation or peripheral stem cell rescue and those with a history of hypocellular marrow, a history of failed stem cell collection, or prior external-beam radiation to more than 25% of the active marrow [7]. The agent is also contraindicated in patients with known type-1 hypersensitivity or anaphylactic reactions to murine proteins or to any component of the product [7].
The agent is administered on an outpatient basis as the radiation exposure risk to family members is in the range of background radiation [1]. The estimated median biological half-life of the agent is 46 to 48 hours [3,6]. The primary route of elimination is via the urine (about 7% eliminated over the first 7 days) [1,3]. No significant radiation is present in ordinary amounts of blood [1]. The median estimated radiation absorbed dose is 3.4 to 5.32 Gy to the liver, 2.0 to 2.6 Gy to the lungs, and 0.1 to 0.38 Gy to the kidneys [2,3,5]. The median estimated dose to the spleen is 8.5 to 9.4 Gy (low doses to the spleen occur when the spleen is not involved with lymphoma) [3]. The estimated tumor absorbed radiation dose is between 15 to 17 Gy [2,3,5]. The tumor-to-normal organ ratio is 7:1 [1].
| Organ | Median radiation dose (in Grays) [5,6] | Range (in Grays) [5] |
| Total body | 0.6 | 0.23-0.79 |
| Red marrow | 0.62-0.97 | 0.18-2.21 |
| Lungs | 2.11-2.16 | 0.94-4.57 |
| Liver | 4.5-5.32 | 2.34-18.56 |
| Kidneys | 0.15-0.23 | 0.03-0.76 |
| Spleen | 7.42-8.48 | 0.76-19.02 |
| Urinary bladder | 0.95 | 0.44-2.70 |
Treatment regimen:
Zevalin is intended as a single course of treatment only [3]. The
treatment regimen previously consisted of two phases which
required 7 to 9 days to complete.The agent can now be administered
without a diagnostic exam.
The diagnostic exam is no longer required, however, patients
still receive an infusion of rituximab 7 days prior to their
therapeutic dose.
1- Diagnostic exam: Patients first undergo a diagnostic exam using
In-111 ibritumomab tiuxetan. A dose of unlabeled rituximab (250
mg/m2) is used to block CD20 binding sites on blood B-cells in the
circulation and in the spleen thereby optimizing biodistribution
of the agent [3]. Rituximab is initially infused at a rate of
50mg/hr [3]. The rate can be increased by 50 mg/hr at 30 minute
intervals up to a maximum rate of 400 mg/hr [3]. Acetaminophen and
diphenhydramine are often given prior to the start of the infusion
to decrease the risk of reaction [3]. In-111 ibritumomab tiuxetan
(5 mCi/1.6 mg antibody in 10 mL) is then given I.V. over 10
minutes [3]. Previously, imaging was performed at 24 hours, 48 to
72 hours, and possibly 90 to 120 hours after administration to
assess the agents biodistribution [3]. Presently, the FDA
recommends only one whole body scan between 48-72 hours following
tracer administration, with subsequent optional scanning to
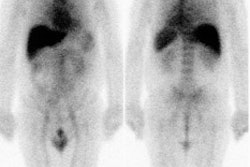
resolve ambiguities [14]. The normal biodistribution on the
initial scan is good uptake in the blood pool areas, moderately
high to high uptake in the normal liver and spleen, and moderately
low to low uptake in the lungs, kidneys, and urinary bladder [3].
Uptake in the blood pool will decrease on later images [7].
Altered biodistribution is suggested when blood-pool activity is
not visualized on the first images, and when lung uptake exceeds
liver activity, renal activity exceeds hepatic activity on
posterior images, and more uptake in the bowel than the liver on
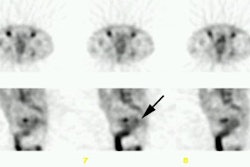
second to third day images [3,7,14]. Increased bone marrow
accumulation of the tracer can be seen in patients with HAMA or
HACA. When altered biodistribution is suspected, whole-body images
should be obtained at 90-120 hours to exclude delayed clearance or
other abnormalities [7]. True altered biodistribution is rare
(approximately 0.6% of patients [14]), but patients with altered
biodistribution should not be treated with 90Y-ibritumomab
tiuxetan
[7]. Once compounded, 111In-ibritumomab tiuxetan has a
shelf-life of 12 hours [7].
2- Therapeutic administration: The agent is dosed by patient body weight and baseline platelet count [1]- unlike Bexar in which the dose is determined by patient specific dosimetry [18]. The dose is 0.4 mCi/kg (14.8 MBq/kg) in patients with platelet counts greater than 150K, and 0.3 mCi/kg (11.1 MBq/kg) if the platelet count is between 100-150K [1,3]. The typical dose range is 21-30 mCi with a maximum administered activity of 32 mCi (although recent studies suggest that patients with excess weight may be insufficiently treated using the predetermined maximum activity [25]) [1,14]. The volume of the dose is usually 4-8 mL [7]. A dose of rituximab (250mg/m2) is administered via slow I.V. infusion (similar to the diagnostic infusion protocol) within 4 hours prior to 90Y-ibritumomab therapy to enhance the biodistribution of the agent by blocking CD20 sites in the peripheral blood and preventing indiscriminate uptake of the agent in the reticuloendothelial system [1]. The agent is given via a slow intravenous infusion over 10 minutes through a low-protein binding 0.22-um filter between the unit dose and the patient [1,7]. The filter should be wet with 0.9% sodium chloride before use and to avoid restricted flow ("air lock") air should not be allowed to pass through the wet filter at any time during syringe set-up or during the infusion [7]. After the infusion, the intravenous tubing should be flushed with 10-20 mL of 0.9% sodium chloride to ensure complete delivery of the dose [7]. Hickman catheters and porta caths may be used for the infusion [7]. Once componded, 90Y-ibritumomab tiuxetan has a shelf-life of 8 hours [7].
For the first 3 to 7 days following treatment, spilled urine and any bodily fluid contaminated material should be cleaned up and flushed down the toilet or placed in a plastic bag in the trash [1,3]. Thorough hand washing after contact with these materials is recommended [1,3].
Toxicity:
The primary adverse event is hemotologic toxicity [3,10]. Grade III/IV neutropenia or thrombocytopenia occurs in roughly two-thirds of patients [3]. Grade IV neutropenia can be seen in 32% of patients and grade IV thrombocytopenia in 5% to 13% of patients [3]. The median absolute neutrophil count nadir is 800, the median platelet nadir is 40,000, and the median hemoglobin nadir is 10.3 g/dL [5]. The median time to nadir is 7 to 9 weeks which is delayed compared to chemotherapy [3]. The median duration of cytopenia is 7 to 35 days [3,10]. The time to complete recovery of blood counts is a median of 99 days [30]. Platelet transfusions can be required in 22% of patients and red cell transfusions in 20% [5]. The risk of hematologic toxicity increases in patients with a greater degree of bone marrow involvement [3,10]. There is a sustained depletion of circulating CD20 B-lymphoctyes following treatment [3]. Recovery begins about 12 weeks after therapy and normal levels by 9 months post-treatment [3]. Patients that have received prior Fludarabine chemotherapy may be more susceptible to the hematologic toxicities of radioimmunotherapy [18].
Neutropenic patients are more prone to develop febrile neutropenia or infections which occur in 7% of treated patients [3,8]. Thrombocytopenic patients can develop lifethreatening hemorrhagic complications (intracranial bleeding occurred in two patients during clinical trials) [3,5].
Following 90Y-ibritumomab tiuxetan treatment, up to one-third of patients may have a subsequent transformation to an aggressive lymphoma [9]. This may be related to a selective cytocidal effect on the low-grade lymphoma cells, thereby giving cells with a more aggressive phenotype a survival advantage [8]. Alternatively, this may represent the natural progression of the disease [8]. Myelodysplasia or acute myelogeneous leukemia occurs in 1.4% of treated patients between 8 to 34 months after therapy [5].
Non-hematologic adverse reactions to the treatment include asthenia, nausea, infection, chills, fever, and abdominal pain [1,7]. Severe allergic reactions to the agent are rare (less than 1% of patients) but include bronchospasm, angioedema, and ARDS [3]. The reactions typically occur 30 to 120 minutes of the first rituximab infusion [3]. Because of the potential for anaphylactic and other hypersensitivity reactions epinephrine, antihistamines, and corticosteroids should be readily available during administration of both rituximab and 90Y-ibritumomab [1]. Patients with a history of hypersensitivity to murine proteins should not receive Zevalin therapy [3].
New human antimouse antibodies (HAMA) develop in 1-2% of treated patients [1,3,5]. Human antichimeric antibodies (HACA) antibodies develop in only 0.5% of patients [5].
Efficacy/Monitoring results of therapy:
Overall response rates of between 50% to 83% have been reported [1-3,8,18,20], with complete response occurring in 8% to 40% [3,5,8,18,20,22]. A recent registry review indicated complete response rates of 73% in patients with follicular lymphoma [30]. Response rates to 90Y-ibritumomab tiuxetan have been shown to be significantly higher when compared to patients that received only rituximab immunotherapy [7,8]. The mean time to disease progress has been report to be 6.8 to 11.2 months [3,5,8]. Higher response rates may be seen in patients with follicular histology [3]. Also- the overall response rates and durations of response are greater when 90Y-ibritumomab tiuxetan is used early in the course of treatment (two or less prior chemotherapies or at first relapse) [14,25,28]. Results have shown that in patients with advanced follicular lymphoma who achieve at least a partial response with induction therapy, subsequent consolidation therapy with a single dose of Zevalin can significantly prolong progression free survival by 2 years, compared with no further treatment [25,31].
The results of the diagnostic scan may also provide prognostic information [22]. A higher rate of complete response has been noted in patients with negative In-111 ibritumomab diagnostic scans (i.e.: no evidence of uptake within sites of bulky adenopathy) [22]. Patients with nodal disease activity identified on the diagnostic exam are less likely to demonstrate a complete response to treatment and are more likely to demonstrate progressive disease (the agent is more effective in patients with non-bulky disease < 5 cm in diameter [28]) [22]. This suggests that patients with bulky disease may require more aggressive management [22].
In trials, Zevalin therapy used for consolidation after first line induction therapy resulted in significantly prolonged progression free survival (approximately 2 years) compared to standard no additional therapy [28]. The treatment also resulted in high partial remission to complete remission conversion rate (77%) [28]. This indicates that Zevalin may have a role in eliminating minimal residual disease [28].
PET/CT may be a more accurate method to assess for complete treatment response as residual masses seen on CT may be considered false-positive for residual disease [20,26]. PET/CT imaging performed as early as 2 months following treatment can be used to identify responders from non-responders [26]. In one study, better overall survival was observed for patients with a reduction in SUV max of 49% or greater from baseline [26]. Early imaging following treatment is beneficial as alternative therapy can be implemented sooner if patients are shown to be non-responders [26].
90Y-ibritumomab tiuxetan treatment does not preclude patients that subsequently relapse from receiving subsequent chemotherapy or autologous stem-cell transplantation [8,9]. Additionally, the treatment has not been shown to effect the efficacy or safety of subsequent therapies [14]. Toxicity with subsequent therapy is similar to that in patients not treated with 90Y-ibritumomab tiuxetan [9].
Elevated baseline LDH and bone marrow involvement have may be associated with a lower overall response rate [18].
Safety:
Because 90Y is a pure beta-emitter, the radiation risk to the patient's family and others is minimal- making it possible to treat patients on an outpatient basis [7]. The calculated potential dose of radiation to others exposed to a treated patient is 1 to 3.5 mrem (0.01-0.079 mSv) [7,19]. For the first 7 days following treatment, patients should wash their hands thoroughly after using the toilet, avoid the transfer of bodily fluids, clean up after spilled urine, and dispose of any materials that are contaminated with bodily fluids [7].
Bexxar (131I-tositumomab):
Tositumomab is an anti-B1 murine monoclonal antibody that binds to the CD20 antigen expressed on the surface of malignant B-lymphocytes [2,4]. The antigen is neither shed nor internalized after antibody binding. 131I-tositumomab is a radioimmunotherapeutic agent which can be used in the treatment of non-Hodgkin's lymphoma. The I-131 gamma emission can be used for documentation of the biodistribution of the agent and to calculate a patient-specific dosage to deliver the desired therapeutic radiation dose [11]. The mean path-length of the beta particle emitted by 131I is 0.4 to 0.8 mm [4,11]. The mean-half life of 131I-tositumomab is 65.2 hours [11]. On average, tumors receive about 10.6 times the mean total body dose [11]. Some authors suggest that Bexar may be preferable to Zevalin for the treatment of patients with lower baseline blood counts or those with less bone marrow reserve due to a less severe decline in platelet counts with this agent [18].
Treatment regimen:
Thyroid blockage is required due to release of 131I from the antibody [2]. A saturated solution of potassium iodide (SSKI or Lugol's) is given from the day before the treatment until 14 days following therapy [4]. The treatment regimen starts by administering unlabeled tositumomab followed by a small dosimetric amount (5 mCi) of the agent to determine individual biologic clearance [4]. Measurements are made immediately following injection and then at two other time points over the next 6 or 7 days [4]. From this information a therapeutic dose is calibrated to deliver the maximum total body dose of 75 cGy (the dose is decreased to 65 cGy in patients with platelet counts of 100-149 K) [4,11]. The dosimetric information is obtained by using the same quantity (in mg) of unlabeled and radiolabeled antibody as will be used for the treatment dose [4]. An indwelling catheter should be used for administration of the treatment [4]. Patients should be premedicated with oral acetaminophen and antihistamine (unless contraindicated) and support equipment should be available to treat anaphylactic reactions (although rare) [4,13]. In order to obtain maximum therapeutic benefit, some authors suggest that larger doses can be administered with planned myeloablation and planned bone marrow or peripheral blood stem cell rescue [21]. In these cases, dosimetry is based on the maximum cummulative dose to critical nonhematopoietic organs (liver, lungs, and kidneys) [21].
For the treatment, patients initially receive a 450 mg infusion of unlabeled Tositumomab (predose) in 50 mL of saline given through a 0.22 microgram filter over 1 hour. The purpose of the pre-dose is to bind to non-tumor B-cells in the circulation, liver, and spleen- thereby protecting them from the therapeutic dose and also allowing the therapeutic radioantibody to bypass binding to these sites and remain bioavailable for tumor binding [4,13]. Splenic update decreases and tumor uptake of the therapeutic dose is higher following a pre-dose regimen [4]. The infusion should be slowed or temporarily stopped of the patient develops fever, rigors, edema, or hypotension [4]. Following the predose, patients receive a 20 minute infusion of 35 mg of 131I-tositumomab in a volume of 30 mL given through a 0.22 microgram filter followed by a 10 minute 30 mL saline flush [4]. Most patients receive a therapeutic dose of 70 to 90 mCi, but the dose can be as high as 150 mCi [4]. Patients do not necessarily need to be hospitalized for the treatment, but they must be able to follow standard guidelines for limiting exposure to other people [4].
Toxicity:
Non-hematologic toxicities are generally mild to moderate and include chills (15%), fever (26-30%), wheezing, nasal congestion/edema, fatigue (32-43%), nausea (25-30%), vomiting (13-15%), infection (13%), pruritus (13%), hypotension (10%), and rash (13%) [11,12].
The most common toxicity is hematologic with a reversible myelosuppression [5,11,12]. In general, toxicity is more severe in patients with more extensive prior therapy [13]. One study has suggested that the amount of time that has elapsed since the patient's last chemotherapy is the most significant predictor for the degree of myelotoxicity (possibly due to incomplete bone marrow recovery following chemotherapy) [29]. Other factors that aid in predicting the degree of toxicity include baseline platelet count and the degree of bone marrow involvement [29]. Cell counts decrease and reach their nadir between 4 to 7 weeks following therapy [4,12] and the nadir can last 20-32 days [13]. The median absolute neutrophil count nadir is between 800 to 1300 cells/mm3, mean platelet nadir 43,000 to 68,000 cells/mm3, and hemoglobin nadir was 10.2 to 11.2 g/dL [5,11,12]. About 21% of patients have platelet counts below 25,000 and between 2% to 11% will have a platelet nadir of less than 10,000 [11,12,13]. Platelet transfusions can be required in 15% to 26% of patients [11,12]. About 2% to 4% to will have an absolute neutrophil count (ANC) of less than 100 cells/mm3 [11] and an ANC of less than 500 cells/mm3 can be seen in about 20% of patients [12]. Blood transfusions can be required in 13-21% of patients and colony-stimulating factor therapy in 13-17% [11]. The median time from treatment to cell count recover is between 2 to 3 months [11,12].
Myelodysplasia can occur following therapy in 3% to 7% of patients, but it is difficult to determine if this is related to prior alkylating agent therapy [12,13].
Free radioiodine can also produce toxicity leading to hypothyroidism [13]. Elevated TSH levels can be found in about 2% of patients following treatment [5], in 9.5% of patients at 2 years after treatment, and in 17.4% at 4 years [13]. Unlike Bexar, hypothyroidism is not associated with Zevalin therapy [13]. Conversely, Bexar would be better tolerated in patients with a history of adverse reactions to rituximab infusions [13].
HAMA formation occurs in 2% to 10% of patients [5,12,13]. However, higher rates of HAMA conversion have been reported (in up to 64% of patients) [5].Conversion to HAMA positivity has not been shown to be associated with altered efficacy of the treatment [12].
Efficacy:
Response rates have been good with reported complete response rates of 16% to 40% and partial response in an additional 25% to 44% of patients [4,5,11,12,18]. The median duration of response is between 6.5 to 9.9 months [11,12]. The median duration of complete response is 6.1 to 19.9 months [11,12]. The duration of response is related to the patients clinical status at the time of treatment (i.e.: size of tumor burden, prior radiation therapy, number of courses of prior chemotherapy) [11,12] and patients have been shown to have overall improved disease response when compared to conventional chemotherapy [12].
In patients that have not previously received chemo or radiation therapy, a response rate of 97% has been reported, with a complete remission in 63% [5]. Elevated baseline LDH and bone marrow involvement have may be associated with a lower overall response rate [18]. Other factors that may be associated with a lower response rate include older age, prior radiotherapy, less than a 75 cGy dose of total body radiation, bulky disease, prior anthracycline containing chemotherapy, and no response to to the last chemotherapy [18].
Safety:
Most patients who are competant to follow instructions can receive therapy on an out-patient basis [13]. Exposure to the general public must be kept below 500 mR [13]. Patients should be instructed to avoid sleeping with other individuals and avoid pregnant people and small children for a week or more [13]. Incontinent or uncooperative patients may not be candidates for treatment [13]. Patients who are unable to comply with radiation safety precautions would be better candidates for Zevalin treatment [13].
Retreatment with 131I-tositumomab is feasible and reasonably effective in HAMA negative patients who have had prior 131I-tositumomab treatment and responded [13].
Estimated radiation-absorbed organs doses with 131I-tositumomab therapy [13]:
The whole body dose is about 0.24 mGy/MBq [Package insert].
| Organ | Median absorbed dose (mGy/MBq) | Range (mGy/MBq) |
| Thryoid | 2.71 | 1.4-6.2 |
| Kidneys | 1.96 | 1.5-2.5 |
| Upper large intestine wall | 1.34 | 0.8-1.7 |
| Lower large intestine wall | 1.30 | 0.8-1.6 |
| Heart wall | 1.25 | 0.5-1.8 |
| Spleen | 1.14 | 0.7-5.4 |
| Testes | 0.83 | 0.3-1.3 |
| Liver | 0.82 | 0.6-1.3 |
| Lungs | 0.79 | 0.5-1.1 |
| Red marrow | 0.65 | 0.5-1.1 |
| Stomach wall | 0.40 | 0.2-0.8 |
REFERENCES:
(1) J Nucl Med 2002; Wagner HN, et al. Administration guidelines for radioimmunotherapy of non-Hodgkin's lymphoma with 90Y-labeled anti-CD20 monocloncal antibody. 43: 267-272
(2) J Nucl Med 2002; Goldenberg DM. Targeted therapy of cancer with radiolabeled antibodies. 43: 693-713
(3) J Nucl Med 2002; Zevalin (Ibritumomab tiuxetan). A promising new regimen for treatment of NHL. 43: 1-8 (DrugLink Advertisement)
(4) J Nucl Med Tech 2002; Seldin DW. Techniques for using Bexxar for the treatment of non-Hodgkin's lymphoma. 30: 109-114
(5) J Nucl Med 2002; Juweid ME. Radioimmunotherapy of B-cell non-Hodgkin's lymphoma: from clinical trials to clinical practice. 43: 1507-1529
(6) J Nucl Med 2003; Wiseman GA, et al. Radiation dosimetry results and safety correlations from 90Y-ibritumomab tiuxetan radioimmunotherapy for relapsed of refractory non-Hodgkin's lymphoma: combined data from 4 clinical trials. 44: 465-474
(7) J Nucl Med Technol 2003; Fink-Bennett DM, Thomas K. 90Y-ibritumomab tiuxetan in the treatment of relapsed or refractory B-cell non-Hodgkin's lymphoma. 31: 61-68
(8) J Clin Oncol 2002; Witzig TE, et al. Randomized controlled trial of Yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin's lymphoma. 20: 2453-2463
(9) J Clin Oncol 2002; Ansell SM, et al. Subsequent chemotherapy regimens are well tolerated after radioimmunotherapy with 90Y-ibritumomab tiuxetan for non-Hodgkin's lymphoma. 20: 3885-3890
(10) J Clin Oncol 2003; Witzig TE, et al. Safety of Yttrium-90 ibritumomab tiuxetan radioimmunotherapy for relapsed low-grade, follicular, or transformed non-Hodgkin's lymphoma. 21: 1263-1270
(11) J Clin Oncol 2000; Vose JM, et al. Multicenter phase II study of iodine-131 tositumomab for chemotherapy-relapsed/refractory low-grade and transformed low-grade B-cell non-hodgkin's lymphoma. 18: 1316-1323
(12) J Clin Oncol 2001; Kaminski MS, et al. Pivotal study of iodine I-131 tositumomab for chemotherapy-refractory low-grade or transformed low-grade B-cell non-Hodgkin's lymphomas. 19: 3918-3928
(13) J Nucl Med 2005; Sharkey RM, Goldenberg DM. Perspectives on cancer therapy with radiolabeled monoclonal antibodies. 46: 115S-127S
(14) J Nucl Med 2005; Conti PS, et al. The role of imaging with 111In-ibritumomab tiuxetan in the ibritumomab tiuxetan (Zevalin) regimen: results from a Zevalin imaging registry. 46: 1812-1818
(15) J Nucl Med 2006; Reilly RM. Radioimmunotherapy of solid tumors: the promise of pretargeting strategies using bispecific antibodies and radiolabeled haptens. 47: 196-199
(16) J Nucl Med 2006; Hicke BJ, et al. Tumor targeting by an aptamer. 47: 668-678
(17) Semin Nucl Med 1995; Wiseman GA, Kvols LK. Therapy of neuroendocrine tumors with radiolabeled MIBG and somatostatin analogues. 25: 272-278
(18) J Nucl Med 2007; Jacene HA, et al. Comparison of 90Y-ibritumomab tiuxetan and 131I-tositumomab in clinical practice. 48: 1767-1776
(19) J Nucl Med 2007; Gulec SA, Siegel JA. Posttherapy radiation safety considerations in radiomicrosphere treatment with 90Y microspheres. 48: 2080-2086
(20) Radiology 2008; Ulaner GA, et al. B-cell non-Hodgkin lymphoma: PET/CT evaluation after 90Y-ibritumomab tiuxetan radioimmunotherapy- initial experience. 246: 895-902
(21) J Nucl Med 2008; Rajendran JG, et al. Myeloablative 131I-tositumomab radioimmunotherapy in treating non-Hodgkin's lymphoma: comparison of dosimetry based on whole-body retention and dose to critical organ receiving highest dose. 49: 837-844
(22) J Nucl Med 2008; Iagaru A, et al 90Y-Ibritumomab therapy in refractory non-Hodgkin's lymphoma: obervations from 111In-Ibritumomab pretreatment imaging. 49: 1809-1812
(23) J Nucl Med 2009; Jacene HA, et al. 18F-FDG PET/CT for monitoring the response of lymphoma to radioimmunotherapy. 50: 8-17
(24) J Nucl Med 2009; Fisher DR, et al. MIRD dose estimate report No. 20: radiation absorbed-dose estimates for 111In- and 90Y-ibritumomab tiuxetan. 50: 644-652
(25) J Nucl Med 2009; Delaloye AB, et al. Dosimetry of 90Y-ibritumomab tiuxetan as consolidation of first remission in advanced-stage follicular lymphoma: results from the international phase 3 first-line indolent trial. 50: 1837-1843
(26) Radiology 2010; Storto G, et al. Assessment of metabolic response to radioimmunotherapy with 90Y-ibritumomab tiuxetan in patients with relapsed or refractory B-cell non-Hodgkin lymphoma. 254: 245-252
(27) J Nucl Med 2010; Gulec SA, et al. Hepatic structural dosimetry in 90Y-microsphere treatment: a monte carlo modeling approach based on lobular microanatomy. 51: 301-310
(28) J Clin Oncol 2008; Morschhauser F, et al. Phase III trial of consolidtion therapy with Yttrium-90-ibritumomab tiuxetan compared with no additonal therapy after first remission in advanced follicular lymphoma. 26: 5156-5164
(29) J Nucl Med 2010; Baechler S, et al. Predicting hematologic
toxicity in patients undergoing radioimmunotherapy with 90Y-ibritumomab
tiuxetan
or 131I-tositumomab. 51: 1878-1884
(30) J Nucl Med 2011; Hohloch K, et al. Radioimmunotherapy
confersw long-term survival to lymphoma patients with acceptable
toxicity: registry analysis by the International
Radioimmunotherapy Network. 52: 1354-1360
(31) J Clin Oncol 2008; Morschhauser F, et al. Phase III trial of
consolidation therapy with yttrium-90-ibritumomab tiuxetan
compared with non additional therapy after first remission in
advanced follocilar lymphoma. 32: 5156-5164
(32) J Nucl Med 2013; Hobbs RF, et al. Radiobiologic optimization
of combination radiopharmaceutical therapy applied to
myeloablative treatment of non-Hodgkin lymphoma. 54: 1535-1542
(33) J Nucl Med 2016; Paprottka KJ, et al. Reduced periprocedural
analgesia after replacement of water for injection with glucose 5%
solution as the infusion medium for 90Y-resin
microspheres. 57: 1679-1684