Adrenergic Nervous System of the Heart
Physiology&
Pharmacology
The sympathetic and parasympathetic innervation of the heart plays a major role in the regulation of cardiac function [14]. Adrenergic fibers that innervate the heart originate in the left and right stellate ganglia [9]. The left stellate innervates the right ventricle, whereas the right stellate innervates the anterior and lateral portions of the heart. The adrenergic fibers travel in the subendocardium following the coronary vessels [9]. At a cardiac level, sympathetic activation results in an increased heart rate (chronotropic effect), augmented contractility (inotropic effect), and enhanced atrioventricular conduction [9].
Sympathetic nervous system dysfunction plays a role in heart
failure [18]. Excessive activity of the sympathetic nervous
system is a major contributor to heart failure progression, by
increaing cardiac work, promoting myocardial fibrosis, and
causing down-regulation of post-synaptic adrenergic receptors
[42]. Noreponephrine (NE) is produced and stored in vesicles in
presynaptic sympathetic nerve terminals [39]. In response to
stimuli, the vesicles are released into the synaptic space with
free NE binding to post-synaptic myocyte receptors producing the
desired cardiac effect [39]. To control the response, there is a
transporter protein mediated, sodium, energy dependent process
(uptake 1) by which free NE is taken back up into presynaptic
terminals for storage or catabolic disposal [39]. Some NE is
also taken up by non-neuronal post-synaptic cells (uptake 2)
[39]. In patients with heart failure, both increased neuronal
release of norepinephrine and
decreased efficiency of NE uptake contribute to increased
cardiac adrenergic drive [18]. Significant reduction in
mortality in heart failure patients can be achieved with the use
of beta- and alpha-adrenoreceptor
blocking drugs [18]. Systemic biomarkers such as B-type
natriuretic peptide (BNP) or N-terminal pro-B-type natriuretic
peptide (NT-proBNP) are released in response to myocardial
stretch and these markers are significantly associated with
sudden cardiac death and ventricular arrhythmias [51].
Metaiodobenzylguanidine (MIBG) is a
guanethidine analog that mimics the
neuronal transport and storage of norepinephrine
[26]. I-123 MIBG has been used to study the cardiac sympathetic
nervous system because of its high cardiac uptake
(unfortunately, I123 MIBG is not FDA approved for cardiac
imaging and therefore not reimbursable [41]). Non-neuronal
activity clears rapidly from the heart between 15 and 60
minutes. In the heart, MIBG is taken up and store by the
postganglionic, presynaptic nerve
endings [21,29]. Similar to NE, there
are two mechanisms for MIBG uptake in adrenergic tissues: a
presynaptic type I neuronal uptake system and a extraneuronal type 2 uptake system (a
minor role in uptake related to a carrier-facilitated process
and diffusion into the myocytes)
[26,29]. As with norepinephrine, I-123 MIBG uptake is
predominantly mediated (70%) via the ATP energy dependent, type
I uptake mechanism [9,26]. After
depolarization, MIBG is released into the synaptic cleft, like norepinephrine, but it is not further
metabolized [21]. MIBG stored within the neuron is also not
further metabolized (it is not a substrate for monoamine oxidase [26,39]) allowing it to
accumulate to concentrations that permit imaging [39]. Only
about 10% of the dose is altered after several days, primarily
by deiodination. The agent produces
no measurable pharmacologic effects. Uptake of I-123 is usually
homogeneous within the myocardium, although uptake may become
more heterogeneous or decrease within the inferior (men) or
lateral (woman) wall as the patient ages [1,2].
Tracer washout also increases with increasing age [2].I-123 MIBG
must compete for uptake with the excess norepinephrine that
accumulates in the synaptic cleft of heart failure patients
which reduces the amount of I-123 MIBG that accumulates in the
presynaptic nerve terminal [53].
11C-meta-hydroxy-ephedrine
(HED) is a PET agent that can be used for neuronal imaging
[33]. The agent has higher uptake selectivity compared to MIBG
and is better in differntiating
between innervated and denervated
myocardium [33]. Patients with CHF have been found to have lower
retention rates of 11C-HED compared to healthy
subjects [47].
Technique:
Patients should fast for 6 hours prior to the exam [13].
Thyroid uptake is blocked by the oral administration of 500 mg
potassium perchlorate (Lugols solution) or a saturated solution
of potassium iodide given 30 minutes before injection of the
radiotracer [20]. Other authors recommend giving 1mg of
potassium iodide from one day before to 1 day after the exam to
block thyroid uptake [30]. Patients are injected at rest [33].
Certain medications and drugs can interfere with catecholamine
and I-123 uptake [33,43,53]. Cocaine and various tricyclic
antidepressants are strong inhibitors of the noepinephrine
uptake-1 transporter and effectively block cardiac uptake of
I-123 MIBG [53]. Sympathicomimetics (ephedrine, pseudoephedrine,
isoproterenol) and reserpine depelete norepinephrine storage
granules which can interfere with I-123 storage in the
presynaptic nerve terminal [53]. Ganglionic blockers and
clonidine block sympathetic nerve activity and alter MIBG
concentration in the heart. Drugs used to improve LV function in
heart failure patients can also improve MIBG uptake and include
beta-blockers (carvedilol, metoprolol, and bisoprolol),
angiotensin-converting enzyme inhibitors, angiotensin receptor
blockers (candesartan), and the aldosterone inhibitor
spironolactone [53]. Opiods, antipsychotics (phenothiazines),
the cardiovascular agent bretylium, and some calcium channel
blockers can also affect MIBG uptake [33,43]. Drugs which block
norepinephrine uptake or deplete norepinephrine stores
(including cocaine and over the counter cold medications) can
decrease MIBG uptake and should be stopped for 5 biologic
half-lives whenever medically feasible [53]. Other authors
suggest they should be held for 24 hours prior to adminsitration of the tracer [33,43].
The beta-blocker labetalol also has signifncant alpha-blocking
activity and can inhibit MIBG uptake [54]. Standard heart
failure medications such as beta-blockers, angiotensin
converting enzyme inhibitors (ACE-I), and/or angiotensin
receptor blockers (ARBs) do not need to be withheld [43]. Foods
containing vanillin and catecholamine-like compounds (chocolate
and blue cheese) should also be avoided [43].
Breast feeding should be stopped for 48 hours after I-123 MIBG
injection [53]. The agent is largely excreted by the kidneys and
patients with severe renal dysfunction may have increased
radiation exposure and decreased image quality [53].
Hypersensitivity reactions have occurred following I-123 MIBG
administration [53].
The dose used for the exam is 3-5 mCi (up to 10 mCi) of I-123 MIBG given over 1 minute
(the higher dose may be required for patients with severe
cardiac dysfunction if SPECT images are to be obtained)
[8,9,13,33,43]. The effective dose from an administered activity
of 10 mCi is 4.8-5.07 mSv [42,53]. The urinary bladder is the
critical organ for I-123 MIBG, but in this same article, they
also indicate that the thyroid is the critical organ [53].
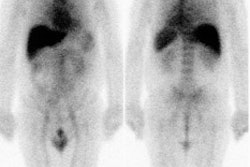
Early (10-15 minute post injection) and delayed (4 hour) planar and SPECT imaging is performed. The neuronal accumulation of MIBG reaches its maximum at 4 hours after injection (hence the delayed image represents actual neuronal uptake as opposed to interstitial uptake on the early images) [26,33,55]. A low-energy parallel hole collimator may used for imaging [20], however, other authors recommend using a medium energy or I-123 collimator in order to minimize noise from scatter from some higher energy I-123 emissions (more than 400 keV) which can affect the H/M ratio (septal penetration by the higher energy photons causes contamination of mediastinal counts by lung activity leading to an underestimation of the H/M ratio [45]) [35,43,45,59]. A 20% window is used and centered over the 159 keV I-123 photon peak [9,20]. If a low-medium energy collimator is used, a 15% energy windo is recommended [45]. An anterior planar image of the chest is acquired for 5 to 10 minutes prior to initiation of SPECT imaging using a 128x128 matrix (some centers also acquire a planar LAO image [9]). Planar images are limited by superimposition of thoracic structures that can also demonstrate MIBG uptake (such as the luns and liver) as well as superimposition of different myocardial segments [29]. Tomographic images are performed to overcome these limitations [29]. SPECT images are obtained every 6 degrees for 30-40 seconds in a 64x64 matrix for a 180 degree rotation (RAO to LPO). Unfortunately, if global myocardial uptake of MIBG is severely reduced, it can be difficult to acquire tomographic images of sufficient quality [29].
Findings:
In healthy subjects, MIBG uptake is slightly lower in the inferior wall (likely due to attenuation, but it has also been suggested to be a physiologic finding relating to vagal tone [29]), apex, and septum [14,29]. It has also been reported that myocardial MIBG uptake decreases with age in adults- particularly in the later decades of life (patients over 60 years of age and therefore, myocardial MIBG uptake has to be corrected for age) [26]. Reduced myocardial uptake of MIBG is seen in association with most diseases that result in cardiac injury [31] and can also be seen in association with cardiotoxicity related to chemotherapy [36].
Image interpretation consists of assessment of global tracer uptake on planar images, tracer washout between early and delayed planar images, and regional uptake on tomographic images [33]. Tracer activity on early imaging is dependent primarily on blood flow and is felt to reflect both the extravesicular and intravesicular accumulation of the tracer [36]. The extravesicular concentration of MIBG decreases rapidly, while the intravesicular concentration remains relatively constant [36]. Hence, delayed images reflect the adrenergic neuron terminal concentration [36].
Cardiac MIBG uptake is semiquantified
by calculating a heart-to-mediastinum
ratio on the planar images [15]. The H/M ratio reflects receptor
density and poortrays both the integrity of presynaptic nerve
terminals and uptake 1 function [39]. The anterior projection
seems to be the preferred projection for quantification as it
provides the lowest variation and highest resolution [19].
Various methods have been described. In one method a 7 x 7 pixel
ROI is placed over the cardiac region and another 7 x 7 pixel
ROI over the midline of the upper mediastinal
area in the the region of lowest activity
(making sure to avoid activity in the thyroid gland)
[13,53]. In another method, left ventricular activity is
measured by manual drawn regions of interest surrounding the
entire myocardium (excluding ventricular blood pool) [15]. A
separate 7 x 7 or 20 x 20 pixel region of interest is placed
over the upper mediastinum. The
heart-mediastinum ratio is
calculated without background subtraction as mean (or average)
counts per pixel over the entire left ventricle divided by mean
(or average) counts per pixel in the upper mediastinum
[9,15,29,55].
A heart-to-mediastinum ratio of
greater than 1.8 is considered normal [9]. Other authors report
a normal HMR as 2.2 +/- 0.3, and a ration of less than 1.6 as
abnormal [33,39,43]. A decreased H/M ratio signifies reduced
cardiac adrenergic receptor density [39]. Some studies suggest
that a regional defect score is superior to the global
heart-to-mediastinum ratio for the prediction of arrhythmic
events- implying that regional heterogeneity may be more
important than global downregulation for the development of
arrhythmia [37].
Another measurement that is calculated is the washout rate
[29]- a ratio of cardiac uptake between early and delayed images
[37]. The washout rate (WR), compensated for tracer decay, is
thought to reflect turnover of catecholamines
and thus sympathetic drive/tone (a measure of the ability of the
myocardium to retain MIBG) [29,33,37,39].
The
clearance
rate
from
the
myocardium
(washout
rate)
is
calculated
by: (Initial myocardial MIBG uptake - Delayed myocardial MIBG
uptake *1.21/ Initial MIBG uptake) x 100 [13]. The factor 1.21
is multiplied by the delayed value to correct for I-123 decay
[43]. The washout rate between early and delayed images should
be less than 10% [9]. Worsening heart failure is associated with
a greater MIBG washout rate, often greater than 27% [39].
Non-uniform soft tissue attenuation over the chest can cause
variations in the measurements obtained [9]. Note- other authors
indicate that the wash out rate is defined as: [H/Mearly
- H/Mlate]/ [H/Mearly] x 100% [53].
In healthy subjects, there is low within subject variability in
I-123 MIBG uptake on both planar (about 5%) and SPECT (about 5%)
imaging [19]. Assuming this is also true for patients with
cardiac disease, the effects of therapeutic interventions can be
monitored using I-123 MIBG imaging [19]. Note: Increased lung
uptake of I-123 MIBG is associated with a better prognosis, in
contrast to increased lung uptake of thallium on MPI [53].
In cardiomyopathies/heart failure:
Cardiac MIBG uptake is decreased in patients with congestive
heart failure, cardiomyopathies,
and ventricular arrhythmias [26]. In heart failure, sympathetic
activity initially increases as manifested by the release of NE
in the synaptic cleft [48]. The upregulated neurotransmitter
release eventually overwhelms the NE transporter 1 (NET-1)
process that then leads to spilling of excess NE into the
circulation [48]. As the HF syndrome progresses, there is
diminished presynaptic function due to loss of neurons and
downregulation of NET-1 (this can be imaged as decreased uptake
of MIBG) [48].
In patients with heart failure the assessment of sympathetic
activity has important prognostic implications regarding risk
stratificaiton and patient survival and will result in better
therapy and outcome [10,32,41].
Alterations in the cardiac sympathetic nervous system clearly
play a role in the development and progression of congestive heart failure (CHF) [13,15]. Altered autonomic function is also
responsible for ventricular arrhythmias or sudden cardiac death
[25]. Decreased cardiac output related to left ventricular
dysfunction results in a baro-receptor
mediated
increased
in sympathetic tone and circulating norepinephrine
levels in an effort to compensate for the decreased cardiac
output [15,33]. The consequences of
chronically increased sympathetic tone include increased left
ventricular afterload, worsening LV
function, and progressive CHF [15]. The hyperactivity of the
sympathetic nervous system in patients with chronic CHF leads to
a downregulation in and
desensitization of myocardial beta-adrenergic receptors [29,40].
It has been shown that in CHF patients, the postsynaptic
beta1-adrenoreceptor density is reduced and the concentration of
inhibiting Gi-alpha proteins is
elevated [13]. Cardiac noradrenaline
turnover is also increased [13]. It is thought that with
progressive heart failure, there is reduced uptake of NE into
presymaptic cardiac nerve storage vesicles via the NE uptake-1
transporter [42]. Chronic NE over-exposure of post-synaptic
beta-adrenergic receptors on the cardiomyocyte results in
beta-receptor desensitization and eventual catoblism and loss of
beta-receptors [42]. Beta-adrenergic blocking agents can aid in
blocking sympathetic over-stimulation associated with CHF and
I-123 MIBG imaging can be used to evaluate the effectiveness of
the treatment [15,32]. Other
physiologic factors that also play a role in CHF include a
decrease in renal blood flow that leads to acceleration of the
renin-angiotensin system (with resultant fluid retention and
exacerbation of heart failure) and also impaired hematopoiesis
[40].
Cardiac MIBG uptake is generally diffusely decreased in patients with dilated cardiomyopathies and in patients with CHF (defects in patients with ischemic cardiomyopathies tend to be more severe in necrotic segments) [11,14]. In these patients, low MIBG uptake is associated with an increased risk for cardiac death [9,11,14,15]. Decreased MIBG uptake on delayed images is closely related to the degree of LV dysfunction in patients with heart failure and also correlates with the level of decreased exercise capacity [8,16]. The poor tracer uptake is likely reflective of underlying cardiac autonomic dysfunction (areas of denervation hypersensitivity) which may place patients at higher risk for arrhythmogenesis [11,35]. The delayed H/M ratio has been reported to be the best predictor for survival in patients with CHF and reduced cardiac function [23]. [52]. In patients with CHF, a normal HMR predicts a <1% yearly risk of cardiac death, while a decreased HMR predicts a poor prognosis [33]. In one study of CHF patients, a HMR<1.2 was associated with a 12 month survival of only 40% [33]. In another study of CHF patients with LVEF
Tracer washout rates have also been studied. Patients with dilated cardiomyopathies typically have accelerated washout rates of MIBG of greater than 25% between early and delayed images (normal less than 10%) [9]. A washout rate of greater than 50% is also associated with an increased risk for cardiac death [9,11] (other quote greater than 27% in chronic CHF patients being associated with a significant increased risk for sudden death (35% cardiac death rate) [29]). The percent change in washout rate between serial exams may also provide information regarding increased risk for cardiac or sudden death (greater than or equal to -5% is associated with an increased risk) [28]. Increased washout can also be seen in other conditions including myocardial hypertrophy and diabetic hearts [11]. MIBG uptake ratios may also help to predict a good response to alpha-blocker therapy in patients with dilated cardiomyopathies [9].
Cardiac resynchronization therapy (CRT) has been shown to be beneficial in patients with advanced chronic heart failure (LVEF <35%) and a QRS duration greater than 120 milliseconds [22,24]. CRT can improve clinical manifestations and quality of life, reduce hospitalizations for CHF, reduce complications, and risk of death (increase survival) [22,24]. Unfortunately, between 20-30% of patients do not respond to CRT [24]. Lower MIBG uptake (H/M ratio below 1.36) is associated with a higher likelihood for lack of response to CRT [22]. The lower MIBG uptake may reflect hearts with more severe myocardial damage that are less likely to respond to CRT [22]. Following successful CRT intervention, there is improved cardiac uptake of MIBG [22]. The presence of left ventricular dyssynchrony also suggests patients that are more likely to respond to CRT [24]. Phase image analysis from gated cardiac examinations can be used to evaluate for the presence of LV dyssynchrony [24]. Extensive LV scarring - particularly when present in the region in which the LV pacing lead is positioned (typically the posterolateral region)- can also result in a decrease likelihood for response to CRT (extensive scarring is predictive of lack of CRT response with a sensitivity of 83% and a specificity of 74%) [24]. This suggests that improvement in LV function is prohibited in the presence of extensive scar tissue [24].
Patients with hypertrophic cardiomyopathies
(HCM) also demonstrate derangements in sympathetic activity
[12]. HCM is an inherited cardiac muscle disease that is related
to a mutation in the genes that encode proteins in the sarcomere [12]. Charateristics
of the disorder include disproportionate left ventricular
hypertrophy and diastolic dysfunction [12]. Delayed MIBG images
demonstrate poor, heterogeneous cardiac tracer retention and
increased washout rates [12,29]. In
patients with HCM, MIBG washout is significantly higher in
patients with ventricular tachycardia (VT) compared to those
without VT [29].
Abnormal 123I-MIBG tracer uptake can also be seen
on SPECT imaging of patients with arrhythmogenic right
ventricular dysplasia- typically with defects in the anterior,
septal, and inferior walls (with normal perfusion to these areas
on perfusion imaging) [38]. In ARVD patients, the presence of
abnormal MIBG SPECT imaging is associated with a significantly
increased risk for ventricular tachyarrhythmia (up to 88% of
patients) [38]. Reduced tracer uptake is likely a reflection of
underlying autonomic dysfunction which places these patients at
an increased risk for dysrhythmia and sudden death [38].
In Monitoring Heart Failure
Response to Therpy:
Beta blockers, angiotensin converting enzyme inhibitors,
angiotensin receptor blockers, and aldosterone inhibitors have
improved outcomes for heart failure patients [42]. Beta blockers
have been shown to reduce cardiac work, up-regulate
post-synaptic adrenergic recpetors, and reduce the risk of death
in patients with chronic heart failure [42]. Other physiologic
factors that also play a role in CHF include a decrease in renal
blood flow that leads to acceleration of the renin-angiotensin
system (with resultant fluid retention and exacerbation of heart
failure). Inhibition of the renin-angiotensin system can be
achieved by blocking the conversion of angiotensin I to
angiotensin II (ACE inhibitors) or by blcoking angiotensin
receptors (angiotensin receptor blockers) [42]. The use of ACE
inhibitors has been shown to improve outcome and mortality in
patients with LV dysfunction [42].
Improvement in MIBG uptake following initiation of beta-blocker therapy may predict which patients are more likely to respond to this form of therapy- even prior to documented LV function improvement [14,42,59]. Patients with preserved HMR on neurohormonal inhibitor medication have improved 5 year survival and a decreased mortality rate [59]. Patients that fail to demonstrate an improved HMR following 6 months of optimal medical therapy are at an increased risk for cardiac death [33].
In heart transplantation:
The surgical procedure of heart transplantation causes autonomic denervation of the donor (allograft) heart [7]. Immediately after heart transplantation no activity is detected in the myocardium [14]. Total denervation persists for at least 12 months after transplantation [14]. Regional reappearance of sympathetic nerve fibers occurs in the transplanted heart over time [7]. MIBG uptake indicating partial sympathetic reinnervation can be shown in 40% of patients 3 to 5 years after transplantation [7]. Serial MIBG studies show that reinnervation begins from the anterolateral base of the heart and spreads towards the apex [9,14]. With reinnervation, patchy MIBG uptake is seen primarily in the anterior, anterolateral, and septal regions. MIBG uptake is usually not apparent in the posterior or inferior myocardial regions, except for basal posterior localization [9]. Complete reinnnervation of the heart is not seen- even up to 12 years after transplantation [9].
In myocardial infarction/ischemia:
Sympathetic nerve fibers are more susceptible to oxygen
deprivation than myocytes and take
longer to recover [29,33]. Uptake of
I-123 MIBG is decreased in areas of acute and chronic ischemia
[9]. Regional denervation of the
heart in the post-ischemic myocardium may persist for 15 days or
longer following an ischemic event [27]. Sympathetic denervation has also been shown in
patients with stable angina in the absence of myocardial
infarction presenting as MIBG defects with preserved perfusion
[29].
Myocardial infarction destroys the myocardium and the nervous
tissue in the myocardium. However, in the early post-infarction
period, the MIBG defect is typically larger than the area of infarcted myocardium (a perfusion-innervation mismatch) [9,14]. This is because infarction can
result in destruction of proximal neurons which supplied innervation to distal areas in which
myocardial tissue is still viable (perfused
and viable, but denervated
myocardium) [14,33]. As a result, a
defect identified on MIBG scintigraphy
following infarction, may be more extensive than the actual area
of infarction as demonstrated by thallium.
As an infarct heals, changes in tissue composition,
repolarization disruption, and autonomic dysfunction can lead to
an increased risk for arrhythmia [39]. There is evidence that
both global and regional sympathetic denervation (anatomic loss
of sympathetic nerves following infarction) or dysinnervation
(sympathetic dysfunction or stunning) predispose patients to
ventricular arrhythmias [39]. Patient's that demonstrate a
perfusion-MIBG mismatch (perfused,
but decreased MIBG activity) have electrophysiologic
abnormalities that can predispose to lethal arrhythmias [33]. Denervated, but viable myocardium has
been shown to be supersensitive to catecholamines
and this may explain an increased risk for arrhythmogenicity
in certain patients following MI [14,29].
Reinnnervation to these peri-infarct regions can be demonstrated
by reappearance of MIBG uptake usually within 14 weeks following
the cardiac event [9]. Unfortunately, reinnervation
may be incomplete [9].
On C-11 HED evaluation, every 1% increase in volume of
denervated myocardium has been shown to be associated with a
5.7% increase risk for sudden cardiac arrest [56]. Patients in
the highest tertile of denervation had a sudden cardiac arrest
rate of 6.7% per year [56].
In patients with ventricular tachyarrhythmias:
Electrophysiologic instability is
an important trigger of cardiac arrhythmias and is modulated by
autonomic function [34]. It has been demonstrated that scar
tissue may serve as a substrate for ventricular arrhythmia (VA)-
particularly the border zone surrounding a scar that consist of
a mixture of both viable tissue and scar (and mIBG uptake in the
border zone may predict recurrent VA) [50].
Abnormally decreased I-123 MIBG uptake can be seen in patients
with ventricular tachyarrhythmias
and is a powerful predictor of recurrent arrhythmic events and
an inferior prognosis [30,34,51]. In
heart failure patients, MIBG imaging provides incremental risk
stratificaiton for the occurence of arrhythmic events
(particularly for patients with HMR < 1.6) [49,51,52]. The
risk for developing life-threatening arrhythmia and cardiac
death is significantly lower for a HMR ≥ 1.6 [51] and in the
ADMIRE-HEX study, no patient with a HMR of at least 1.8
experienced a fatal or potentially fatal arrhythmic event,
compared to an event rate of 6-10% for patients with a HMR <
1.6 [52]. The presence of stress perfusion defects in
non-ischemic cardiomyopathy patients also increases the risk for
arrhythmic events (particular in patients with a HMR < 1.6
and a SRS >8) [51].
Regional cardiac sympathetic denervation can be found on SPECT imaging in up to 67% of patients with ventricular tachycardia (compared to 8% of control patients) [39]. I-123 MIBG imaging can effectively indicate which patients are likely to benefit from ICD [43]. The presence of resting perfusion defects is associated with an increased arrhythmic risk in patients with non-ischemic cardiomyopathy and heart failure (EF < 35%) with a decreased H/M ratio on I-123 MIBG imaging [46]. In one study, a H/M ratio < 1.54 was associated with an increased incidence of ICD discharges and on SPECT imaging, patients who had ICD discharges had more extensive autonomic/perfusion mismatches [43]. Unfortunately, due to globally decreased tracer uptake in heart failure patients, SPECT imaging can be challenging [51]. However, even the presence of myocardial scar on SPECT perfusion imaging has been shown to further risk stratify patients with low HMR < 1.6 [56]. In the ADMIRE-HF cohort, patients with a HMR < 1.6, but a summed rest score ≤8 had fewer episodes of sustained VT, resuscitated sudden cardiac arrest, and appropriate ICD therapies when compared to those with SRS > 8 (3.9% vs 11.9%) [56].
Other settings of abnormal myocardial innervation:
Parkinson's disease: Reduction of 123I-MIBG uptake
or (11C-hydroxyephedrine (HED) on PET imaging) in the
heart (due to cardiac sympathetic neuronal loss) is considered a
specific finding for idiopathic Parkinson's disease (IPD)
without autonomic failure and can be used to differentiate it
from other parkinsonian syndromes
[29,57]. 11C-HED PET studies have demonstrated
significant heterogeneity of cardiac denervation in IPD patients
[57]. A regional pattern of denervation has been described
preferentially involving the inferior and lateral LV walls, and
relatively sparing the anterior and septal walls [57]. On short
term (2 years) followup, IPD patients with baseline abnormal
scans demonstrate progressive decline in cardiac sympathic
neuronal integrity, but patients with normal HED scans have
demonstrated no subsequent denervation and this may represent a
protected phenotype [57]. Another study of IPD patients, also
demonstrated a subgroup with early disease that had normal or
mildly abnormal MIBG scans- this group of patients was
characterized as female-dominant, young onset, slow progression
in motor dysfunction, and preserved cognitive function [58].
A significant decrease in MIBG uptake is seen in the inferior and lateral segments in hypertensive patients with cardiac hypertrophy [29].
Cardiac sympathetic denervation/autonomic dysfunction in diabetic patients can also be evaluated with MIBG [29]. Decreased MIBG uptake in diabetics is associated with an increased mortality rate [29].
PET Adrenergic Imaging:
Norepinephrine (NE) is stored in neuronal vesicles and
synthesized from tyrosine- tyrosine is converted to
dihydroxyphenylalanine (DOPA) by rate-limiting tyrosine
hydroxylase; DOPA is converted to dopamine by DOPA-decarboxylase
which is actively transported into sotrage vesicles by vesicular
monoamine transporter (VMAT); within the vesicle dopamine is
converted to NE by dopamine beta-hydroxylase; some NE is further
converted to epinephrine by
phenylethanolamine-N-methyltransferase [44]. A sympathetic nerve
impulse leads to docking of vesicles to the axonal membrane
where its contents are released into the sympathetic cleft [44].
Over 80% of NE utilized by the heart is synthesized in the
cardiac sympathetic neurons which necessitates the active
recapture of the neurotranmitter from the synapse [44]. The
active recapture is mediated by the NE reuptake transporter
(uptake-1) which returns synaptic NE to the neuronal cytosol for
packaging into vesicles or degradation by monoamine oxydase and
catechol-O-methyltransferase
[44]. Uptake-1 is saturable, can be blocked by reuptake
inhibitors (such as cocaine and desipramine), and is dependent
on ATP and sodium [44]. A small portion of synaptic NE undergoes
transport via the uptake-2 pathway [44]. Uptake-2 is
non-saturable and is not dependent on ATP or sodium [44].
Corticosteroids and clonidine have been shown to inhibit
uptake-2 [44].
C11-hydroxyephedrine (HED-
a norepinephrine analog) has been used for mapping cardiac
sympathetic activity [29]. The agent is resistant to metabolism
by monoamine oxidase and
catecholamine O-methyltransferase,
has a high affinity for the uptake 1 transporter mechanism, and
is partially packaged into vesicles by vesicular monoamine
transporter (VMAT) [29,51]. HED is believed to undergo
continuous relaease and reuptake by
sympathetic neurons [29]. The distribution of the tracer in the
myocardium is normally homogeneous without the decreased
activity in the inferior wall noted on SPECT MIBG imaging [29].
A marker for the quantification of C11-HED uptake is the
retention index which is defined as the ratio between activity
in the myocardium and th integral of the arterial blood-time
activity curve [51]. The C11-HED global retention index closely
correlates with the late I123-MIBG HMR [51]. The volume of
viable denervated myocardium on C11-HED imaging shows a
significant association with the time to sudden cardiac arrest
[51].
[F18] 6-fluorodopamine may also be useful for cardiac imaging and the agent has a longer half-life [29]. The tracer is accumulated mainly via the presynaptic uptake 1 mechanism and then sequestered into sotrage vesicles and beta-hydroxylated to fluoroepinephrine [29].
Conditions/drugs
which decrease cardiac MIBG uptake
- Pheochromocytomas/Elevated
circulating levels of norepinephrine
which competes with MIBG for the type I uptake system
- After eating
- Administration of
yohimbine (due to increased
adrenergic activity)
- Administration of
tricyclic antidepressants (imipramine, desipramine)
- produce a moderate reduction in cardiac MIBG uptake
- Administration of
sympathomimetics
(pseudoephedrine or phenylpropanoloamine)
- Administration of ladetalol- strong inhibitory effect on cellular uptake of MIBG [54]
- Administration of
slective serotonin reuptake inhibitors or
serotonin-norepinephrnie reuptake inhibitors
- Administration of reserpine- produces norepinephrine depletion through the irreversible inactivation of vesicular monoamine transporter, thereby preventing NE storage in vesicles and increasing the rate of enzymatic degradation [54]
- Cocaine use-
cocaine and other monoamines have an inhibitory effect on
cardiac MIBG uptake [54]
- Cardiovascular
autonomic neuropathy of diabetes mellitus: Cardiovascular
autonomic neuropathy is a serious complication of diabetes
and the prevalence can be as high as 20-30% of patients with
non-insulin dependent diabetes (NIDDM) [21]. Decreased
cardiac MIBG uptake in diabetic patients is associated with
an increased mortality [9,21].
Improvement in glycemic control
has been shown to result in partial restoration of
sympathetic innervation [14].
- Following cardiac
transplantation (within 1 year, beyond this time about 50%
of patients may demonstrate uptake indicative of re-innervation)
- Following
ascending aortic aneurysm surgical repair- most likely the
result of mechanical damage to the cardiopulmonary nerves
surrounding the aorta [10].
- Following
chemotherapy with doxorubicin: Decreased myocardial MIBG
uptake can be seen following doxorubicin therapy, with
limited morphologic damage [9]. Decreased MIBG uptake
follows a dose dependent decline with about 25% of patients
demonstrating some decrease in MIBG uptake at cumulative
doses of 240-300 mg/m2 [9]. Decreased MIBG uptake precedes
deterioration of ejection fraction [9,14].
Evidence
of sympathetic damage can be used to select patients at risk
of severe functional impairment and who may benefit from cardioprotective agents or changes
in the schedule of antineoplastic
drugs [9].
- Congestive heart
failure secondary to pressure or volume overload
- Dilated cardiomyopathy- washout is also
increased in these patients [6]
- Acute Myocarditis [6]
- Myocardial
infarction [6]
- Sympathetic nerve
destruction by stellate ganglionectomy
- Epicardial phenol application, and Shy-Drager syndrome.
- Parkinson's
disease [10,14]. MIBG
abnormalities observed in Parkinson's patients may be due to
postgangliotic sympathetic
dysfunction and has been correlated with severity and length
of disease [14].
- LV hypertrophy due
to essential hypertension [14]. MIBG abnormality is mainly
observed in the inferior and lateral walls and the degree of
abnormality correlates with the severity of hypertrophy
[14].
Conditions
which increase cardiac MIBG uptake
- Administration of
clonidine (an alpha-2 antagonist
which slows nerve traffic)
- Administration of
amiodarone [54]
- Heart failure
secondary to coronary artery disease
REFERENCES:
(1) J Nucl Med 1995; Tsuchimochi S, et al. Age and gender differences in normal myocardial adrenergic neuronal function evaluated by iodine-123-MIBG imaging. 36: 969-974
(2) J Nucl Med 1998; Sakata K, et al. Physiologic fluctuation of the human left ventricle sympathetic nervous system assessed by Iodine-123-MIBG. 39: 1667-1671
(3) Nucl Med Annual 1993; Sisson JC. The adrenergic nervous system of the heart and nuclear medicine. Ed. Freeman LM. Raven Press, NY.: 233-249 (No abstract available)
(4) J Nucl Med 1995; Mantysaari M, et al. Myocardial sympathetic nervous dysfunction detected with iodine-123-MIBG is associated with low heart rate variability after myocardial infarction. 36: 956-61
(5) J Nucl Med 1995; Glowniack JV. Cardiac studies with metaiodobenzylguanidine: a critique of methods and interpretation of results. 36: 2133-37 (No abstract available)
(6) J Nucl Med 1998; Agostini D, et al. Impariment of cardiac neuronal function in acute myocarditis: Iodine-123-MIBG scontography study. 39: 1841-1844
(7) J Nucl Med 2001; Odaka K, et al. Reappearance of cardiac presynaptic nerve terminal s in the transplanted heart: Correlation between PET using 11C-hydroxyephedrine and invasively measured norepineephrine release. 42: 1011-1016
(8) J Nucl Med 2001; Zhao C, et al. Comparison of cardiac sympathetic nervous function with left ventricular function and perfusion in cardiomopathies by 123I-MIBG SPECT and 99mTc-Tetrofosmin electrocardiographically gated SPECT. 42: 1017-1024
(9) J Nucl Med 2001; Carrio I. Cardiac neurotransmission imaging. 42: 1062-1076
(10) J Nucl Med 2001; Momose M, et al. Total and partial cardiac sympathetic denervation after surgical repair of ascending aortic aneurysm. 42: 1346-1350
(11) J Nucl Med 2001; Wakabayashi T, et al. Assessment of underlying etiology and cardiac sympathetic innervation to identify patients at high risk of cardiac death. 42: 1757-1767
(12) J Nucl Med 2002; Shimizu M, et al. Heterogeneity of cardiac sympathetic nerve activity and systolic dysfunction in patients with hypertrophic cardiomyopathy. 43: 15-20
(13) J Nucl Cardiol 2002; Parthenakis FI, et al. Segmental pattern of myocardial sympathetic innervation in idiopathic dilated cardiomyopathy: relationship to regional wall motion and myocardial perfusion abnormalities. 9: 15-22
(14) J Nucl Cardiol 2002; Patel AD, Iskandrian AE. MIBG imaging. 9: 75-94 (No abstract available)
(15) J Nucl Cardiol 2002; Gerson MC, et al. Carvedilol improves left ventricular function in heart failure patients with idiopathic dilated cardiomyopathy and a wide range of sympathetic nervous system function as measured by iodine 123 metaiodobenzylguanidine. 9: 608-615
(16) J Nucl Med 2003; Sato M, et al. Correlation between cardiac norepinephrine overflow during exercise and cardiac 123I-MIBG uptake in patients with chronic heart failure. 44: 1618-1624
(17) J Nucl Med 2003; Inoue Y, et al. Effect of collimator choice on quantitative assessment of cardiac iodine 123 MIBG uptake. 10: 623-32
(18) J Nucl Cardiol 2003; Narula J, Sarkar K. A conceptual paradox of MIBG uptake in heart failure: retention with incontinence. 10: 700-704
(19) J Nucl Cardiol 2004; Somsen GA, et al. Normal values and within-subject variability of cardiac I-123 MIBG scintigraphy in heathy individuals: implications for clinical studies. 11: 126-133
(20) J Nucl Cardiol 2004; Flotats A, Carrio I. Cardiac neurotransmission SPECT imaging. 11: 587-602
(21) J Nucl Cardiol 2006; Mushtaq N, et al. I-123 metaiodobenzylguanidine imaging in non-insulin-dependent diabetic patients with normal myocardial perfusion scans: new insights into their increased cardiac morbidity and mortality rates. 13: 8-10
(22) J Nucl Cardiol 2007; D'Orio Nishioka SA, et al. Cardiac sympathetic activity pre and post resynchronization therapy evaluated by 123I-MIBG myocardial scinitgraphy. 14: 852-859
(23) J Nucl Med 2007; Kasama S, et al. Additive effects of spironolactone and candesartan on cardiac sympathetic nerve activity and left ventricular remodeling in patients with congestive heart failure. 48: 1993-2000
(24) J Nucl Med 2007; Henneman MM, et al. Nuclear imaging in cardiac resynchronization therapy. 48: 2001-2010
(25) J Nulc Med 2008; Nagahara D, et al. Predicting the need for an implantable cardioverter defibrillator using cardiac metaiodobenzylguanidine activity together with plasma natriretic peptide concentration or left ventricular function. 49: 225-233
(26) J Nucl Cardiol 2008; Chen W, et al. Age-related decrease in cardiopulmonary adrenergic neuronal function in children as assessed by I-123 metaiodobenzylguanidine imaging. 15: 73-79
(27) J Nucl Med 2008; Vesely MR, Dilsizian V. Nuclear cardiac stress testing in the era of molecular medicine. 49: 399-413
(28) J Nucl Med 2008; Kasama S, et al. Prognostic value of serial cardiac 123I-MIBG imaging in patients with stabilized chronic heart failure and reduced left ventricular ejection fraction. 49: 907-914
(29) J Nucl Cardiol 2008; Henneman MM, et al. Cardiac neuronal imaging: application in the evaluation of cardiac disease. 15: 442-55
(30) J Nucl Med 2009; Akutsu Y, et al. The significance of cardiac sympathetic nervous system abnormality in the long-term prognosis of patients with a history of ventricular tachyarrhythmia. 50: 61-67
(31) J Nucl Cardiol 2009; Jacobson AF, et al. 123I-mIBG scintigraphy to predict risk for adverse cardiac outcomes in heart failure patients: design of two prospective multicenter international trials. 16: 113-121
(32) J Nucl Cardiol 2009; Flotats A, Carrio I. Radionuclide noninvasive evaluation of heart failure beyond left ventricular function assessment. 16: 304-315
(33) J Nucl Cardiol 2010; Yong S, Travin MI. Radionuclide imaging of cardiac autonomic innervation. 17: 655-666
(34) J Nucl Med 2010; Nishisato K, et al. Impaired cardiac sympathetic innervation and myocardial perfusion are related to lethal arrhythmia: quantification of cariac tracers in patients with ICDs. 51: 1241-1249
(35) J Nucl Cardiol 2011; Tamaki N, et al. Novel iodinated tracers, MIBG and BMIPP for nuclear cardiology. 18: 135-143
(36) J Nucl Med 2011; de Geus-Oei LF, et al. Scintigraphic techniques for early
detection of cancer treatment-induced cardiotoxicity.
52: 560-571
(37) J Nucl Med 2011; Nuklearmedizin K, et al. Imaging targets
of the sympathetic nervous system of the heart: translational
considerations. 52: 1167-1170
(38) J Nucl Med 2011; Paul M, et al. Cardiac sympathetic
dysfunction in genotypes patients with arrhythmogenic right
ventricular cardiomyopathy and risk of recurrent ventricular
tachyarrhythmias. 52: 1559-1565
(39) J Nucl Cardiol 2012; Kelesidis I, Travin MI. Use of
cardiac radionuclide imaging to identify patients at risk for
arrhythmic sudden cardiac death. 19: 42-52
(40) J Nucl Med 2012; Doi T, et al. Cardiac mortality
assessment improved by evaluation of cardiac sympathetic nerve
activity in combination with hemoglobin and kidney function in
chronic heart failure patients. 53: 731-740
(41) J Nucl Cardiol 2012; Travin MI, Kamalakkannan G. A key
role for nuclear cardiac imaging in evaluating and managing
patients with heart failure. 19: 879-882
(42) J Nucl Cardiol 2012; Waqar F, et al. What will be the role
of I-123 MIBG in improving the outcome of medically treated
heart failure patients? 19: 1198-1205
(43) J Nucl Cardiol 2013; Travin MI, et al. Cardiac autonomic
imaging with SPECT tracers. 20: 128-143
(44) J Nucl Cardiol 2013; Thackeray JT, Bengel FM. Assessment
of cardiac autonomic neuronal function using PET imaging. 20:
150-165
(45) J Nucl Med 2013; Inoue Y, et al. Acquisition protocols and
correction methods for estimation of the heart-to-mediastinum
ratio in 123I-metaiodobenzylguanidine cardiac
sympathetic imaging. 54: 707-713
(46) J Nucl Cardiol 2013; Sood N, et al Resting perfusion
MPI-SPECT combined with cardiac 123I-mIBG
sympathetic innervation imaging improves prediction of
arrhythmic events in non-ischemic cardiomyopathy patients:
sub-study from the ADMIRE-HF trial. 20: 813-820
(47) J Nucl Cardiol 2013; Ademaw N, Salerno M. PET/MRI: current
state of the art and future potential for cardiovascular
applications. 20: 976-989
(48) J Nucl Cardiol 2013; Gulati V, et al. The role of
radionuclide imaging in heart failure. 20: 1173-1183
(49) J Nucl Cardiol 2014; Al Badarin FJ, et al. The utility of
ADMIRE-HF risk score in predicting serious arrhythmic events in
heart failure patients: incremental prognostic benefit of
cardiac 123I-mIBG scintigraphy. 21: 756-762
(50) J Nucl Cardiol 2014; Zhou Y, et al. I-123 mIBG and Tc-99m
myocardial SPECT imaging to predict inducibility of ventricular
arrhythmia on electrophysiology testing: a retrospective
analysis. 21: 913-920
(51) J Nucl Cardiol 2014; Wollenweber T, Bengel FM. Molecular
iaging to rpedict ventricular arrhythmia in heart failure. 21:
1096-1109
(52) J Nucl Med 2015; 123I-MIBG imaging for
prediction of mortality and potentially fatal events in heart
failure: the ADMIRE-HEX study. 56: 1011-1018
(53) J Nucl Cardiol 2015; Soman P, et al. I-123 MIBG cardiac
imaging. 22: 677-685
(54) J Nucl Cardiol 2015; Jacobson AF, Travin MI. Impact of
medications on mIBG uptake, with specific attention to the
heart: comprehensive review of the literature. 22: 980-993
(55) J Nucl Cardiol 2016; Henzlova M, et al. ASNC imaging
guidelines for SPECT nuclear cardiology procedures: stress,
protocols, and tracers. 23: 606-639
(56) J Nucl Cardiol 2016; Malhotra S, Canty JM.
Life-threatening ventricular arrhythmias: current role of
imaging in diagnosis and risk assessment. 23: 1322-1334
(57) J Nucl Med 2017; Wong KK, et al. 2-year natural decline of
cardiac sympathetic innervation in idiopathic Parkinson disease
studied with 11C-hydroyephedrine PET. 58: 326-331
(58) J Neurol Neurosurg Psychiatry 2015; Tsujikawa K, et al.
Chronological changes of 123I-MIBG myocardial
scintigraphy and clinical features of Parkinson's disease. 86:
945-951
(59) J Nucl Cardiol 2017; Nakajima K, et al. Cardiac sympathetic nervous system imaging with 123I-meta-iodobenzylguanidine: perspectives from Japan and Europe. 24: 952-960