Neuroimaging advances are playing a critical role in the understanding of movement disorders. PET in particular is proving invaluable for assessing a wide range of clinical issues, including treatment-related complications of Parkinson’s disease, preclinical neurodegenerative changes in patients who are gene-positive for Huntington’s disease, and brain abnormalities in asymptomatic carriers of the DYT1 mutation, which causes the most common form of dystonia.
At the 2002 International Congress of Parkinson’s Disease and Movement Disorders in Miami, multiple presenters discussed the role of PET in studying these diseases. They noted that research facilities differ in the level of involvement that their radiologists have in such studies. Radiologists may perform the imaging themselves, or they may interpret studies that have been performed by neurologists.
PET and Parkinson’s disease
Parkinson’s patients often develop fluctuations in their response to dopaminergic therapy and go on to develop dyskinesias. PET scans have shown that fluctuators have a lower uptake of 18FDG, and that such patients have an altered pattern of dopamine release after taking oral levodopa.
 |
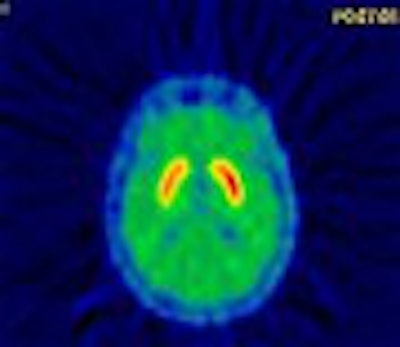
| These images show PET scans performed with 11Craclopride in the same patient, at open baseline (no injection) and after injection of a saline placebo. The reduction in binding represents release of dopamine in response to placebo. Stoessl and colleagues first reported these results in the article, "Expectation and Dopamine Release: Mechanism of the Placebo Effect in Parkinson’s Disease" (Science, August 10, 2001, Vol. 293:5532 pp. 1164-1166). Image courtesy of Dr. A. Jon Stoessl. |
"PET studies are helping us gain some more insight regarding why treatment-related complications occur in Parkinson’s disease," said Dr. A. Jon Stoessl, professor of neurology at the University of British Columbia in Vancouver. "At this point we are not using imaging as a diagnostic tool, though. We’re getting interesting results from PET studies that suggest complications may be related to increased dopamine turnover." Stoessl also noted that the results of a PET scan could indicate an increase in dopamine turnover, which might be a preclinical manifestation of Parkinson’s disease.
 |
| This graph shows the relationship between FDG measured by PET and the number of dopamine (tyrosine hydroxylase) neurons in the substantia nigra measured at later post-mortem. Previous data (labeled as old data) are expanded upon (new data). Image courtesy of Edith McGeer and Dr. A. Jon Stoessl. |
In recent work, Stoessl and colleagues have used the radiotracer raclopride due to its ability to bind to D2 receptors. "If an individual has sufficient dopamine, less raclopride will bind to the receptors," he said. "Therefore, we can try to estimate progression by changes in the availability of raclopride."
Four-hour raclopride scans have documented fluctuations in levodopa availability. In images obtained before and after the administration of levodopa, Stoessl and colleagues were able to document the treatment effect fading four hours after treatment in patients with fluctuations.
 |
| PET scans of one Parkinson’s disease patient using three different tracers. Image courtesy of Dr. A. Jon Stoessl. |
"Even if you scan patients with stable response to medication, we see two different patterns emerging," he said. "In one group you see a slow and steady increase in the availability of dopamine. In others, you see an early and sharp increase followed by a return to baseline within four hours, as demonstrated by changes in raclopride binding. People with sharp increases typically develop fluctuations and motor complications within three years."
Preclinical Huntington’s patients
Raclopride PET imaging may also be useful in tracking the course of preclinical Huntington’s disease, according to preliminary data, which shows an approximate nine-year lapse between the onset of neurodegeneration and the presence of clinical symptoms.
"We do not yet know when the brain starts to degenerate, but we do know that by the time people have clinical signs and symptoms, considerable degeneration has taken place, particularly in the basal ganglia," said Dr. Klaus Leenders, professor of neurology at University Hospital in Groningen, the Netherlands.
He noted that radiotracer studies have been able to identify metabolic defects in preclinical patients because the tracer is sensitive to medium spiny projection neurons, which are present in the striatum and contain a high number of D2 receptors. These neurons are reduced in the early stages of Huntington’s disease, and raclopride PET studies can easily document this loss, Leenders said.
Most PET studies to date have been performed in patients with established Huntington’s disease, Leenders added.
 |
| The first results using cerebral uptake of radiotracers measured with PET in patients with overt Huntington’s disease, HD mutant gene carriers, gene-negative siblings, and healthy control subjects. Raclopride was used to determine dopamine D2 receptor binding in striatal regions, and FDG was used to measure regional energy metabolism. Here the values of the caudate nucleus are given. The gene carriers were clinically non-symptomatic. This small group of gene carriers showed metabolic values partly below normal and partly within normal range. Image courtesy of Dr. Klaus Leenders. |
"In our work, we look at presymptomatic people because we want to know whether we can predict when the neurons will show injury," he said. "If our preliminary data are right -- that there are a certain number of years that lapse consistently before clinical symptoms occur -- we might be able to identify mutant gene-positive people before they’re symptomatic. If so, it seems that they would be the right population for experimental therapy."
Leenders and colleagues have an ongoing study to determine whether there is a uniform pattern of preclinical neurodegeneration. "We have finished the baseline data with 40 people," he said. "In the first phase, half of the people were still biochemically normal, and half had neurodegenerative changes that were outside the range of gene-negative. We are now in the second phase, a two-year follow-up, in which we are comparing the gene load, atrophy, and ranking within the group of people who are gene-positive. We will also follow these patients at four and six years."
Brain abnormalities and the dystonia gene
A recent FDG-PET study documented abnormal brain function among non-manifesting carriers of the mutated DYT1 gene. This mutation is associated with the most common form of progressive, early-onset dystonia, now called DYT1 dystonia, which typically progresses from a focal disorder with symptoms in one limb to debilitating, generalized dystonia.
According to Dr. David Eidelberg, professor of neurology and neurosurgery at New York University School of Medicine in New York City, PET studies show an abnormal resting metabolic brain network in asymptomatic carriers of the mutation compared to normal controls. Eidelberg’s team recruited seven right-handed carriers and seven right-handed age-matched controls, and scanned the subjects while they were moving their dominant hand on a digitizing tablet.
All of the subjects performed two motor tasks. The first entailed reaching movements in a predictable counterclockwise order. The second involved the presentation of targets in a repeating pattern, with subjects synchronizing target acquisition so that they were learning a sequence and anticipating the correct target.
 |
Brain regions in which activation differences were recorded during motor sequence learning in DYT1 carriers and controls. In gene carriers, increased learning-related activation was present in the right pre-SMA (top) and in the anterior lateral cerebellum (bottom). Images courtesy of Dr. David Eidelberg.
 |
The investigators found that the two groups did not differ in general motor performance. In the first task the two groups’ results were similar; however, in the second task, the carriers showed reduced learning performance. This result correlated with an increase in learning-related activation of the left prefrontal cortex and lateral cerebellum. In normal subjects, sequential learning is typically correlated with the activation of a brain network involving the posterior parietal regions.
"The FDG-PET images of the two groups are different," Eidelberg said. "These scans showed abnormalities among the carriers in the way the brain activates itself during motor tasks. Even though they’re clinically normal, an abnormality can be detected in complex motor behavior. When the carriers of this gene learn things with their hands, their movements are organized in a problematic sequence. In such people, the brain is not organized normally, even though they are neurologically normal from a clinical standpoint."
Ironically, in families with the DYT1 mutation, non-manifesting carriers consider themselves lucky. "Such people carry the gene but are grateful that they’re not affected," Eidelberg said. "They have relatives with severe dystonia. We used to think of DYT1 dystonia as a disease only affecting those with the symptoms. However, brain imaging shows that even non-manifesting carriers of the mutation have profound abnormalities."
By Paula MoyerAuntMinnie.com contributing writer
March 4, 2003
Related Reading
MRI advances diagnosis of Hallervorden-Spatz syndrome, January 29, 2003
Imaging agent aids diagnosis of Parkinson’s, December 4, 2002
New tracers, technologies propel clinical applications in PET, June 14, 2002
PET, SPECT confirm: In Parkinson's, dopamine agonists work better than levodopa, May 20, 2002
PET predicts future cognitive decline, June 26, 2001
Copyright © 2003 AuntMinnie.com