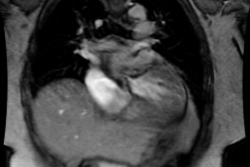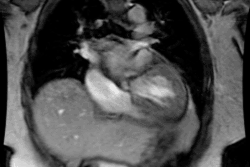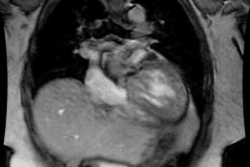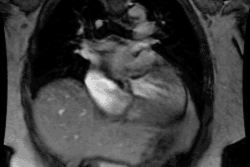
Tetrology of Fallot:
Clinical:
Tetrology accounts for 10-11% of cases of congenital heart disease [4]. Tetrology is the most common cyanotic congenital heart disease associated with decreased pulmonary vascularity and it is the most common cause of cyanotic congenital heart disease beyond the neonatal period (representing 75% of all cyanotic heart disease). The degree of cyanosis is dependent upon the severity of right ventricular outflow tract obstruction and associated pulmonic stenosis. Cyanosis is not detected in most patients until at least 3-4mo. of age, but it may not present until early childhood if the obstruction is mild.Tetrology results from a single defect (an anterior malalignment of the conal septum [4]) and consists of:
1- VSD (High), typically a large membranous defect (anterior conal septum below right aortic cusp)
2- Overriding Aorta: The aorta overrides the VSD and in combination with pulmonic stenosis results in right ventricular outflow tract obstruction
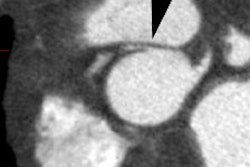
3- Pulmonic Stenosis: The stenosis is subvalvular (infundibular) in 90% of cases, but occasionally may be both valvular and infundibular. The pulmonic stenosis exacerbates the degree of right to left shunting. If the stenosis is mild, however, patients may only be cyanotic following exercise. The pulmonic valve may be stenotic or bicuspid. About 40% of patients have an associated stenosis of the origin of the left or right pulmonary artery.
4- Right Ventricular Hypertrophy. Pentalogy of Fallot consists of the above plus an ASD.
Patients with more severe pulmonic stenosis will present as
cyanotic
neonates. Older children typically present with episodic syncope
and frequent
squatting (which increases systemic resistance to divert more
blood into
the pulm. circulation). The amount of cyanosis increases with
exercise,
hot weather, and feeding. Clubbing and polycythemia may be seen,
but CHF
does not usually occur. Other cardiac anomalies associated with
Tetrology
include a right aortic arch which is noted in 25% of patients. Of
these,
90% will have mirror imaging branching of the great vessels, and
the other
10% an aberrant left subclavian. Other associated anomalies
include peripheral
pulmonary artery coarctation, unilateral pulmonary artery
atresia/stenosis
(esp. left), and coronary artery anomalies (in up to 36% of
patients) such
as an anomalous LAD arising from the right coronary artery.
Associated
non-cardiac anomalies include tracheo-esophageal fistula, Down's
syndrome,
and skeletal anomalies (scoliosis).
Less common forms of condition include TOF with pulmonary artery
atresia and aortopulmonary collaterals, and rarely TOF with absent
pulmonary valve or TOF with an AV canal defect [10]. Tetrology of
Fallot with absent pulmonary
valve syndrome is an important form of the disorder. It is
characterized
by a large VSD, a small pulmonary annulus, and nubbins of valve
tissue
instead of leaflets which produces severe pulmonic stenosis. There
is resultant
massive dilatation of the pulmonary arteries which compress the
airways-
the cyanosis in these cases is related to the airway compression
rather
than right-to-left intracardiac shunting [3].
Surgical treatment of TOF:
Both palliative and corrective surgical repair have resulted in
improved
long term survival in tetrology of Fallot patients. The
Blalock-Taussig
shunt is a palliative systemic to pulmonary artery anastomotic
shunt from the subclavian
artery (opposite the arch) to the corresponding pulmonary artery
to increase
pulmonary blood flow [5,6]. The ipsilateral vertebral artery is
also ligated
to prevent subclavian steal syndrome. Drawbacks of the procedure
were possible abnormal growth and strength of the ipsilateral arm
[7]. Unilateral rib notching involving
the upper ribs on the side of the anastamosis may develop
secondary to
enlargement of collateral vessels supplying arterial blood to the
affected
upper extremity. The shunt may also be created using a synthetic
graft
which is referred to as the modified Blalock-Taussig shunt [5,7].
Benefits of
the modified procedure are that it can be performed on either side
and
distal flow in the subclavian artery is maintained. Unfortunately,
because
the shunt conduit size is fixed, children may outgrow the
anastamosis as
soon as 2 years after the procedure. Other complications include
pulmonary artery stenosis (14% of cases) and shunt narrowing (50%
of cases) usually at the anastomosis [8]. Asymmetric pulmonary
blood flow is
common following either procedure, with greater flow to the
ipsilateral
lung. Reduced diastolic blood flow in the coronary circulation can
be associated with the use of a Blalock-Taussig shunt [5].
A Sano shunt consists of an extracardiac allograft valved conduit inserted directly from the right ventricle to the pulmonary arteries [5]. The Potts operation (side-to-side anastomosis of the descending aorta to the left pulmonary artery) and the Waterston operation (side-to-side anatomosis of the ascending aorta to the right pulmonary artery) are also corrective procedures, but eventually lead to pulmonary hypertension, LV volume overload, and pulmonary artery tortuosity [6].
Definitive repair had previously been postponed until age 5-7
years, but is now usually performed in the first year of life
(often in the first 6 months) [5,9]. The procedure consists
of patching/closing the VSD and relieving the pulmonary outflow
obstruction by
reconstructing the RV outflow tract (removing the obstructing
muscle) or establishing a separate conduit from the RV to the main
pulmonary artery [5,10].
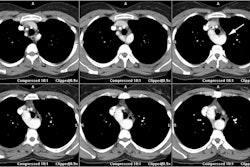
On follow-up, calcification in the conduit suggests restenosis of
the outflow
tract. Definitive repair performed after this time is associated
with impaired
RV function in response to exercise. Repair after the age of 40
years is
associated with a 17% surgical mortality.
Complete repair of TOF with PA atresia may be preceded by
palliative shunts and "unifocalization" of aortopulmonary
collaterals into a common vessel [10]. The timing of VSD closure
in pulmonary atresia depends on the growth of the reconstructed PA
and resulting RV pressures [10]. Residual and recurrent proximal
and distal PA stenoses may result in RV hypertension after VSD
closure [10]. Catheterization procedures with pulmonary balloon
angioplasty or stent placement (or both) are common in these
patients before and after complete repair with VSD closure [10].
In TOF with absent pulmonary valve, PA plication to relieve
airway compression is often performed at the time of VSD closure
in patients with respiratory symptoms [10]. For both TOF with
pulmonary atresia and TOF with absent pulmonary valve, a RV to PA
conduit is placed to the branch pulmonary arteries [10].
Patients with TOF and AV canal defect usually undergo
simultaneous repair of both lesions and are at risk of
postoperative LV outflow obstruction from left-sided AV valve
tissue [10].
X-ray:
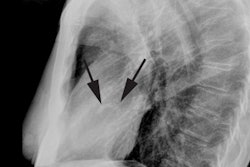
On plain film there is typically decreased pulmonary vascular markings, but the heart and vessels may be normal. The main pulmonary artery segment will be concave (post-stenotic dilatation is seen with valvular, but not with infundibular stenosis). RVH results in elevation of the cardiac apex. Cardiomegaly may be noted, but the heart size can be normal. A right aortic arch is noted in 25% of cases [4]. Classically the heart develops a "wooden shoe" (coeur in sabot) shape due to uplifting of the cardiac apex as a result of right ventricular hypertrophy and concavity of the main pulmonary artery [4].Differential considerations for a concave main pulmonary segment and decreased pulm. blood flow include: Hypoplastic right heart syndrome, transposition of the great vessels and pulmonic stenosis, double outlet RV and pulm. stenosis, single ventricle and pulm. stenosis, and pseudo-truncus (Type IV Truncus- also referred to as Tetrology with pulmonary atresia. As the pulmonary arteries are absent the lungs are supplied by systemic collaterals).
REFERENCES:
(1) J Thorac Imag, 1995, 10: 1-25
(2) J Thorac Imag, 1995, 10: 26-35
(3) Pediatric Clinics of North America 1999; Waldman JD, Wernly JA. Cyanotic congenital heart disease with decreased pulmonary blood flow in children. 46(2): 385-402
(4) Radiographics 2007; Ferguson EC, et al. Classic imaging signs of congenital cardiovascular abnormalities. 27: 1323-1334
(5) AJR 2007; Rodriguez E, et al Postoperative imaging in cyanotic congenital heart diseases: Part I, normal findings. 189: 1353-1360
(6) AJR 2008; Takaki MTT, et al. Nonatherosclerotic cardiovascular findings on MDCT coronary angiography: a selection of abnormalities. 190: 934-946
(7) Radiology 2008; Gaca AM, et al. Repair of congenital heart disease: a primer-part 1. 247: 617-631
(8) AJR 2008; Spevak PJ, et al. Surgically corrected congenital heart disease: utility of 64-MDCT. 191: 854-861
(9) Radiographics 2010; Frank L, et al. Cardiovascular MR imaging
of conotruncal anomalies. 30: 1069-1094
(10) J Cardiovasc Comput Tomogr 2013; Han BK, Lesser J. CT
imaging in congenital heart disease: an approach to imaging and
interpreting complex lesions after surgical intervention for
tetrology of Fallot, transposition of the great arteries, and
single ventricle. 7: 338-353