Alveolar Proteinosis:
View cases of alveolar proteinosis
Clinical:
Pulmonary alveolar proteinosis (PAP) can be understood as a syndrome of altered surfactant homeostasis, leading to a pathologic accumulation of surfactant [6]. Because surfactant homestasis is complex, there are many potential points of disruption- overproduction of phospholipids byt type II pneumocytes, impairment of clearance of phospholipids by macrophages, or both [6]. As a consequence of this disruption, floccular periodic acid-Schiff (PAS) positive proteinaceous and lipid rich material is deposited within the alveoli.
There are two forms of alveolar proteinosis described:
(1) A "primary", autoimmune, or idiopathic form occurring in the absence of an identifiable associated disease or exposure (possibly related to an immunologic disturbance) [9,10]. This form accounts for the vast majority of cases (90%) and these patients have high levels of autoantibodies against granulocyte macrophage colony stimulating factor (GM-CSF) in their blood and tissues, including pulmonary alveoli (unfortunately, titers do not correlate with disease severity) [9,10].
(2) A "secondary" form has also been described, provoked by or associated with another condition. Secondary alveolar proteinosis has been described in association with: 1) infections of the lung; 2) hematologic malignancies and other conditions altering a patients immune status (AIDS or pharmacologic immunosuppression); and 3) exposure to inhaled mineral dusts (silica, aluminum, titanium), toxic fumes, and chemicals (insecticides) [9,11]. The most frequent underlying disorder for secondary PAP is hematologic disease such as myelodysplasic syndrome, acute myeloid leukemia, and chronic myeloid leukemia [10]. Secondary PAP is likely related to a relative deficiency of GM-CSF and to the resulting macrophage dysfunction, thereby compromising surfactant clearance [11]. The prognosis of patients with secondary PAP is linked to the underlying disease [11].
(3) A controversial congenital third form has also been described [9].
Patients generally present between the age of 20-50 years (mean 40 years) with progressive shortness of breath and mild, usually non-productive cough (mean duration 7 months [9]). Weight loss, fatigue, and malaise may also be present. However, up to 30% of patients will be asymptomatic even with florid CXR findings. There is a strong association with tobacco use- about 75% of PAP patients are smokers and within this subgroup males are affected more than females (3:1) [9]. In nonsmokers, there is no sex predilection [9]. Superimposed pneumonia affects about 13% of patients- often opportunisitic such as Nocardia, Candida, Cryptococcus, Aspergillus, etc... [9].
Physical exam findings are usually non-specific, but clubbing (25%) and cyanosis (21%) can be seen. An elevated serum lactate dehydrogenase level is the most common associated laboratory abnormality (82% of patients), but this finding is non-specific [9]. Patients also have elevated levels of lung surfactant proteins A and D in both the serum and BAL fluid. The intrapulmonary shunt fraction while breathing 100% oxygen is typically elevated (average 20%). Most patients have mild to moderate restrictive lung disease with decreases in total lung capacity, forced vital capacity, forced expiratory volume in 1 second, and diffusion capacity for carbon monoxide [6].
The disorder usually follows a variable course with exacerbation's and remissions. Spontaneous improvement or recovery occurs in 25-50% of cases. Previously up to 30% mortality within several years had been reported, but the actual mortality may be less than 10% [4]. Treatment is whole lung bronchopulmonary lavage (WLL) to remove abnormal surfactant which can lead to improved oxygenation as alveoli re-expand [9], but this is not required in all cases (about 2/3's of patients with PAP require WLL within 5 years of diagnosis [9]). After treatment, marked improvement in the disease and radiographic findings are usually apparent [6]. About 80% of patients respond favorably to lung lavage [6] and the mean symptom free interval following WLL is 15 months [9]. Lung transplant can be used in selected cases, but the disorder can recur in the transplanted lung [7,9].
For patients with secondary PAP< treatment consists of removal of the inciting agent [9].
Concomitant superinfection can occur, classically with Nocardia,
but more
recently described with Mycobacerium avium-intracellulare
and pneumocystis
carinii. [1]
Pulmonary fibrosis develops in 1.4% to 20% of patients with PAP
[11].
X-ray:
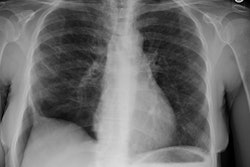
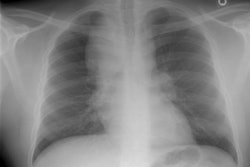
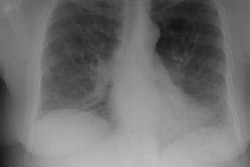
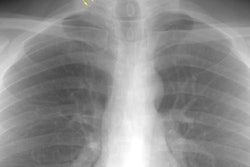
CXR: On CXR there are small acinar nodules which may coalesce to form areas of consolidation- typically in a central, perihilar "bat-wing" configuration which may mimic pulmonary edema, although the heart size is normal and pleural effusions are typically absent. The involvement can be asymmetric (up to 44% of cases [9]) or very rarely unilateral (1% of cases [9]). There is often a notable disparity between the moderate clinical symptomatology of PAP and the more impressive radiographic findings (clinicoradiologic discrepancy) [9]. Lymphadenopathy and pleural effusion are rare (although an effusion can be seen following lavage [6]). Changes typically resolve slowly over weeks to months. Later there is diffuse reticulogranularity and changes consistent with interstitial lung disease.
Computed tomography: HRCT demonstrates patchy geographic areas of ground glass opacification or consolidation and peripheral or central ill-defined nodular opacities. Subpleural sparing is common [10]. Smooth interlobular septal thickening is frequently seen within the areas of ground-glass opacification (up to 75% of cases [9]). The septal thickening produces an underlying polygonal pattern referred to as "crazy paving." The "crazy paving" pattern can also be seen in cases of left heart failure [9], pneumocystis pneumonia [9], alveolar hemorrhage [9], mucinous bronchoalveolar cell carcinoma [8], lipoid pneumonia, ARDS [9], and radiation or drug-induced pneumonitis [5,9]. Diffuse ground-glass opacificaition and absence of subpleural sparing can be seen and are more commonly associated with secondary PAP [10].
REFERENCES:
(1) Chest 1997; Diagnosing pulmonary alveolar proteinosis. A review and an update. Chest 111(2): 460-466 (No abstract available)
(2) AJR 1997; Ground-glass opacity at CT: the ABCs. 169(2): 355-367 (No abstract available)
(3) Chest 1997; Lee KN, et al. Pulmonary alveolar proteinosis: high-resolution CT, chest radiographic, and functional correlations. 111: 989-995
(4) Chest 1998; Goldstein LS, et al. Pulmonary alveolar proteinosis. Clinical features and outcomes. 114: 1357-1362
(5) Radiology 1999; Johkoh T, et al. Crazy-paving appearance at thin-section CT: Spectrum of disease and pathologic findings. 211: 155-160
(6) AJR 2001; Holbert JM, et al. CT features of pulmonary alveolar proteinosis. 176: 1287-1294
(7) Radiology2001; Collins J, et al. Frequency and CT findings of recurrent disease after lung transplantation. 219: 503-509
(8) AJR 1997; High-resolution CT findings of mucinous bronchioloalveolar carcinoma: A case of pseudopulmonary alveolar proteinosis. 168: 99-100 (No abstract available)
(9) Radiographics 2008; From the archives of the AFIP. Pulmonary alveolar proteinosis. Frazier AA, et al. 28: 883-899
(10) Chest 2009; Ishii H, et al. Comparative study of
high-resolution CT findings between autoimmune and secondary
pulmonary alveolar proteinosis. 136: 1348-1355
(11) AJR 2016; Akira M, et al. Pulmonary fibrosis on
high-resolution CT of patients with pulmonary alveolar
proteinosis. 207: 544-551