Pulmonary Arterial Hypertension:
Clinical:
The NIH defines primary pulmonary artery hypertension as a mean
pulmonary artery pressure more than 25 mm Hg at rest (normal 10-20
mm Hg; 21-24 mm Hg is considered abnormal) or greater than 30 mm
Hg with exercise (normal 15 mm Hg) measured during right heart
catheterization [15,18,19,22], with the exclusion of etiologies
for secondary pulmonary hypertension as listed below. The severity
of PAH is catorgorized as: mild = more than 25 mm Hg, but less
than 30 mm Hg; moderate = 30-50 mm Hg; and severe more than 50 mm
Hg [33]. The pulmonary venous wedge pressure should be normal (15
mm Hg or less) when the condition is precapillary, and greater
than 15 mm Hg if the condition is post capillary [22,26].
Pulmonary artery hypertension (PAH) can be divided into primary and secondary etiologies. It is also classified as either pre-capillary (with changes limited to the pulmonary arterial circulation, mainly at the level of the muscular arteries) or post-capillary (with findings located within the pulmonary venous circulation, between the capillary bed and the left atrium- i.e.: disorders which increase pulmonary venous pressure) [18]. Post capillary pulmonary hypertension is further divided into two groups- isolated post capillary pulmonary hypertension if the diastolic pressure gradient (diastolic PCP minus PAWP) is lower than 7 mm Hg and combined post- and pre- capillary hypertension if the diastolic pressure gradient is equal or greater than 7 mm Hg [32].
| PH |
Precapillary PH |
Postcapillary PH |
Mixed PH |
| mPAP > 25 mm Hg |
PCWP | PCWP > 15 mm Hg |
PCWP > 15 mm Hg |
| PVR = 3 WU |
PVR | PVR > 3 WU |
|
| TPG >/= 12-15 mm Hg |
TPG < 12 mm Hg |
TPG >/= 12-15 mm Hg |
PCWP- pulmonary capillary wedge pressure; PVR- pulmonary venous
resistance; TPG- transpulmonary gradient; WU- Wood unit [33].
Pre-capillary causes of PAH include primary pulmonary
hypertension, collagen vascular disease, HIV, long-standing
left-to-right shunt, chronic thromboembolic disease [18].
Post-capillary causes of PAH include pulmonary venoocclusive
disease, mitral valve disease, left heart failure, left atrial
tumor, and mediastinal fibrosis (which can also affect
pre-capillary vessels) [18].
The World Health Organization classifies pulmonary hypertension
(PH) into five
clinical groups- [28,33]
Group 1- Pulmonary arterial hypertension (idiopathic, or associated with connective tissue disease or HIV)
Group 2- PH due to left heart disease
Group 3- PH due to chronic lung disease or hypoxia
Group 4- Chronic thromboembolic PH
Group 5- PH of unclear or multifactoral mechanisms
1- Primary Pulmonary Hypertension (Plexogenic pulmonary arteriopathy):
Primary pulmonary hypertension is a diagnosis of exclusion.
Primary pulmonary hypertension is a rare disorder of unknown etiology which primarily affects females (1.7 to 3:1). The mean age at presentation is 35 years (most common range 20-45 years [20]) and the most common symptoms are dyspnea (60%), fatigue (19%), and syncope or near syncope (13%) [15]. The average delay between the onset of symptoms and the diagnosis of PAH is 2 years [20]. The disorder has an association with autoimmune disorders. Raynaud's phenomena is observed in about 10% of patients with primary PAH, it is more common in women, and it is associated with a worse prognosis. A positive antinuclear antibody test is found in about 30% of affected patients.
A familial form of primary pulmonary hypertension has also been described. It accounts for about 6-10% of cases of primary pulmonary hypertension [8]. The familial form is inherited as an autosomal dominant trait with variable expression and patients typically present at a younger age. The most common physical exam findings include hypoxia, a loud pulmonary component (P2) of the second heart sound, a right sided S3, peripheral edema, elevated jugular venous pulsations, and hepatomegaly as a result of tricuspid regurgitation or right heart failure [15].
Histologically there is a plexogenic pulmonary arteriopathy (characterized by hypertrophy of the smooth muscle cells of the media) and an in situ thrombotic arteriopathy. The pathologic changes are largely confined to the muscular pulmonary arteries (those less than 1 mm in diameter that contain a medial layer of muscle) [14]. Pulmonary hypertension results in right ventricular hypertrophy, followed by dilatation as the demand for oxygen by the RV exceeds the available supply [22]. Dilatation results in tricuspid regurgitation due to incomplete valve closure and subsequently right atrial enlargement [22]. These changes eventually result in decreased cardiac output and right-sided heart failure [22]. Pressures on the left side of the heart (pulmonary capillary wedge pressure) are usually normal until the right heart chamber dilatation compresses the left side.
Without therapy the disorder is typically progressive with death in 2 to 3 years due to progressive right heart failure (median survival 2.8 years and 5-year survival of 34% [20]). With therapy, five year survival rates up to 95% have been reported. Treatment usually consists of vasodilator and anticoagulation drug therapy. The calcium channel blockers nifedipine and diltiazem are the most commonly used vasodilators and they produce a sustained improvement in 25-30% of patients. Epoprostenol (prostacyclin) is a new very effective vasodilator agent with antiplatelet aggregating effects [15]. Epoprostenol inhibits smooth muscle proliferation and may facilitate pulmonary vascular remodeling of occluded arterial beds [15]. The agent is given by continuous infusion via a portable infusion pump and permanent indwelling central line. The agent has been shown to improve pulmonary hemodynamics and exercise tolerance [13]. Inhalation of an aerosolized prostacyclin analogue (iloprost) has also be used for therapy [16]. Anticoagulation is required in patients with primary PAH due to an increased risk for thrombosis and thromboembolism as a result of the sluggish pulmonary blood flow (acute and organizing thrombi are noted in over 50% of cases of primary pulmonary hypertension [9]). Oral anticoagulation has been shown to improve survival in patients with PAH [15].
Patients with primary pulmonary hypertension may be eligible for
lung transplantation. Even patients with markedly depressed RV
function, will improve following transplant. Unfortunately,
mortality rates are significantly higher after transplantation for
patients with primary pulmonary hypertension- one year mortality
is between 30-35%, and three year survival is between 50-60%. The
survival following transplant is slightly worse than that for
aggressive vasodilator therapy [8]. Only patients failing to
improve on medical treatment should be considered for lung
transplant [8]. Recurrence of pulmonary hypertension after
transplant has not been reported. [1]
- Heritable PAH: Heritable PAH is found in 6-10% of patients and
is associated with mutations of the bone morphogenic protein
cascade [29].
2- Secondary Pulmonary Hypertension: Etiologies include:
- Chronic pulmonary disease: COPD (about 50-66% of patients with
severe COPD have some degree of PAH, although it is generally
mild [22,29]), emphysema, cystic fibrosis, end-stage lung. PAH
is reported to have a prevalence as high as 46% in patients with
idiopathic pulmonary fibrosis [20]. However, in patients with
pulmonary fibrosis, dilatation of the main pulmonary artery may
occur in the absence of PAH (thus, the main pulmonary artery
diameter at CT is an unreliable indicator of PAH in this patient
group) [20]. In patients with pulmonary fibrosis, the ratio of
the PA to the ascending aorta is a more reliable marker of PAH
[20]. PH is believed to be the result of hypoxic pulmonary
vasoconstriction and remodeling of the pulmonary vessels [29].
The hypoxic vasoconstriction is initially reversible, but over
time it may become irreversible [29].
- Collagen vascular disease: Scleroderma (particularly in patients with CREST syndrome [15]), SLE (5-10% of patients [23]), mixed connective tissue disease [23], and less commonly, rheumatoid arhtritis [8,23].
- Vascular disease: Congenital heart disease with left to right
shunt [Eisenmenger's syndrome is associated with ASD, VSD, and
PDA], recurrent
thromboembolic
disease (pulmonary hypertension develops in 3.8% of
patients with chronic thromboemboli [20]), vasculitis, primary pulmonary
veno-occlusive disease, fibrosing mediastinitis, pulmonary
capillary hemangiomatosis, pulmonary venous hypertension,
and portal hypertension.
Eisenmenger syndrome: Eisenmengers syndrome occurs when pulmonary vascular resistance approaches or exceeds systemic vascular resistance and a left-to-right shunt becomes bidirectional or reversed [22]. At this point, correction of the shunt is no longer possible and a heart or lung transplant is required [22]. It is calculated that 10% of patients with a VSD of any size may develop Eisenmenger syndrome, compared to 4-6% of subjects with an ASD [20]. The incidence can be up to 50% in patients with large VSD's, and 10% with large ASD's [20]. Eisenmengers syndrome may be associated with massive aneurysmal dilatation of the central pulmonary artery, in situ thrombus formation (that may calcify), and atheromatous calcification of the pulmonary arteries [22].
- Left heart disease: Pulmonary hypertension induced by left
heart disease is the most prevalent form worldwide [29]. It
begins as increased back-pressure with congestion of the
pulmonary capillary resulting in elevated pressures [29].
- Sickle cell disease: PAH affects 6-10% of adult sickle cell
disease patients, likely the result of chronic thromboembolic
disease [31].
- Drugs: The appetite suppressants fenfluramine and dexfenfluramine are most commonly associated with producing pulmonary artery hypertension, but also benflurex [29]. The drugs are inhibitors of serotonin uptake. The risk of PAH increases with increasing duration of use, but PAH can still occur with exposure of only a short duration [10]. Stopping the medication may have a substantial improvement in the condition, and remission may occur.
Other drugs associated with pulmonary arterial hypertension include amphetamines, cocaine, phenylpropanolamine, selective serotonin reuptake inhibitors, and L-trytophan [8,29].
These drugs may also have a contributory effect to the development of pulmonary hypertension in patients with other secondary causes of PAH [10].
- HIV infection: HIV infection is an independent risk factor for the development of pulmonary artery hypertension [8,11]. The exact mechanism for the development of pulmonary hypertension in this group of patients is unclear, but plexogenic pulmonary arteriopathy (similar to primary PAH) is the most common histopathologic finding [11,12,24]. Currently, the estimated incidence of PAH among HIV infected individuals is 0.5% [17,29] which is an increase of 6-25 times the general population [18,27]. There is no sex predilection (although some authors suggest it is more common in men [27]) and the diagnosis of HIV infection precedes the diagnosis of pulmonary hypertension in 94% of cases [11]. There is no correlation between the length of disease or the CD 4 count and the presence of pulmonary hypertension [11,12,15]. More than 79% of patients have CD4 counts greater than 200 at the time of diagnosis [25]. Symptoms include shortness of breath (85%), pedal edema (30%), non-productive cough (19%), and syncope or near syncope (12%). Dilatation of the right heart chambers is the most common echocardiographic finding (98%) [11]. The interval between the diagnosis of HIV disease and the diagnosis of pulmonary hypertension is about 33 months [11]. The median length of time between diagnosis and death is 6 months [11]. Responses to vasodilator agents and antiretroviral therapy is variable and very difficult to measure accurately [11]. Although other authors suggest that HIV associated PAH responds to treatment with prostacyclin epoprostenol and the oral dual-endothelin receptor agonist, bosentan [25]. Confounding factors for secondary pulmonary hypertension can be found in about 18% of cases [11].
- Parasitic embolization: Schistosoma mansoni infection can lead to pulmonary artery hypertension [9]. S. mansoni eggs travel as emboli from porto-systemic collaterals to lodge in the lung where they incite an inflammatory antigenic obliterative endarteritis.
- Pulmonary talcosis [9]- pulmonary talcosis occurs when IV drug
users inject crushed oral medication into peripheral veins and
the talc becomes trapped within the pulmonary arterioles
producing vascular occlusion [22]. On HRCT, there is a fine
diffuse, micronodular pattern and ground-glass attenuaiton, as
well as areas of paraseptal emphysema [22].
Determination of the etiology can greatly aid in patient
management. Patients with chronic thromboembolic disease are now
treated with surgery.
Complications of PAH:
Marked enlargement of the pulmonary artery in patients with PAH
may compress the left main or LAD coronary arteries resulting in
decreased coronary blood flow [34]. The best predictor of left
main coronary artery stenosis equal to or greater than 50% was a
PA diameter equal to or greater than 40 mm [34].
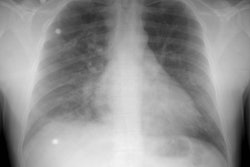
X-ray:
On plain film, the principal radiologic indication of PAH is
disparate enlargement of the central pulmonary arteries compared
to the peripheral branches. Note that central enlargement is
essential to the diagnosis- it is not necessary that the
peripheral vessels be pruned or decreased in number [5]. On the PA
CXR, a right interlobar artery greater than 16 mm in diameter (15
mm in women) at the level of the junction of the right mainstem
bronchus and the SVC is plain film evidence of pulmonary
hypertension. The descending left pulmonary artery should exceed
18 mm [5].Right ventricular dilatation results in a reduced
retrosternal air space on the lateral view [22].
Echocardiography: Echo is an excellent screening tool for PAH
[22]. The reported sensitivity is between 79-100% and specificity
is 68-98% [22].
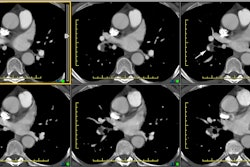
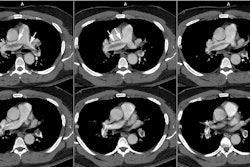
The CT determined diameter of the main pulmonary artery is
measured in the scan plane of the bifurcation just lateral to the
aorta, at a right angle to the vessels long axis [9] 2 cm from the
pulmonary valve [26]. A main pulmonary artery wider than 29 mm on
CT is consistent with PAH [7] (Sensitivity 84-87%; Specificity
75-89%; PPV > 95% [9,20,22,26]). However, it is important to
recognize that a diameter of less than 29 mm does not necessarily
exclude PAH [22]. A pulmonary artery-to-aorta ratio greater than 1
(main pulmonary artery segment larger than the aorta) also has a
strong correlation with PAH (PPV 96%, specificity 92%, sensitivity
70%), especially in patients younger than 50 years of age
[12,20,22,26]. A main PA diameter ≥
29mm and a segmental artery to bronchus ratio greater than 1:1
in three of four pulmonary lobes is highly specific for PAH
(specificity 100%) [20]. There should also be an
abrupt decrease in the caliber of the segmental and sub-segmental
arteries [20]. It should be recognized that older age, male sex,
and higher body surface area are each correlated with larger PA
size and PA ratio decreases with increasing age [34].
Additionally, the PA ratio is not applicable in the case of aortic
dilatation [34].
The Fleishner Society makes the following recommendations [34]:
| CT criteria |
Low risk population for PAH |
Intermediate risk for PAH |
High risk for PAH |
Suspected PAH |
| PA diameter (mm) |
> 34 mm |
> 32 mm |
> 30 mm |
Any size |
| PA-Aorta ratio |
> 1.1 |
> 1.0 |
> 0.9 |
Any value |
Diameters of the left and right pulmonary arteries (upper limit
of normal 16 mm) are less consistent indicators for PAH [18].
Dilatation of the bronchial and nonbronchial systemic arteries is
seen in only 14% of cases of idiopathic PAH and is much more
common in patients with recurrent thromboembolic disease (seen in
up to 73% of cases) [20].
The "egg-and-banana" sign refers to visualization of the main PA
at the same level as the most caudal aspect of the aortic arch -
the finding has a high specificity and PPV for PAH [30].
Other findings on CT which suggest pulmonary hypertension include
heterogeneous lung attenuation (mosaic attenuation) that is higher
centrally, than peripherally. A expiratory image may be necessary
to demonstrate that these findings are not secondary to
emphysematous changes within the lungs. A mosaic attenuation
pattern has been described in pulmonary artery hypertension due to
vascular disease/chronic PE (most commonly), cardiac disease, or
lung disease [2]. The presence of ill-defined centrilobular
opacities (felt to represent cholesterol granulomas or large
plexogenic arterial lesions) has also been described in patients
with primary pulmonary hypertension and in PAH secondary to left
to right shunting [3] and in patients with pulmonary capillary
hemangiomatosis [22,26].
Cardiac signs of PAH include right ventricular hypertrophy, which is defined as a RV wall thickness of more than 4-6 mm, straightening or leftward bowing of the interventricular septum, right ventricular dilatation (a RV-to-LV ratio of more than 1:1, and dilatation of the IVC and hepatic veins with reflux of contrast material due to elevated right heart pressures [22,26]. Pericardial thickening and pericardial effusions can also be seen [6]. The likelihood of finding a pericardial effusion increases with the severity of PAH and its presence implies a worse prognosis [20].
On ventilation-perfusion scintigraphy, perfusion is typically heterogeneous, with small peripheral defects, however, the scan may also be normal. Recently, the presence of reverse mismatches have been described in patients with primary pulmonary hypertension. A reverse mismatch is referred to as an area of lung which has absent or diminished ventilation, but is perfused normally, or to a greater degree than the remainder of the lungs. If extensive, this finding is of great physiologic importance as blood shunted through poorly ventilated lung tissue can be a major cause of hypoxia. This finding is felt to reflect an inadequate hypoxic vasoconstriction reflex and may be related to lack of vessel responsiveness secondary to replacement of medial hypertrophy by intimal hyperplasia in late stages of the disorder. In patients with primary pulmonary hypertension HRCT demonstrates a characteristic mosaic attenuation pattern. Areas of reverse mismatch on the V/Q scan have been shown to correlate with areas of increased attenuation on HRCT [4].
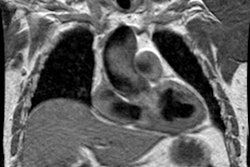
MRI:
Delayed contrast enhanced MR will demonstrate foci of enhancement
in the LV myocardium at the septal RV insertion sites (more
prominent at the base of the heart) in patients with pulmonary
hypertension and the extent of enhancement has been correlated
with increased afterload and inversely correlated with RV
performance [21,22]. The finding is likely related to increased
mechanical stress in these areas with resultant scarring [22].
Phase contrast imaging can be used to determine the average flow
velocity in the main pulmonary artery [22]. A mean average
velocity in the main pulmonary artery of less than 11.7 cm/sec has
a sensitivity of about 93% and a specificity of about 82% for the
detection of PAH [22].
REFERENCES:
(1) N Engl J Med 1997; Rubin LJ. Primary pulmonary hypertension. 336(2), 111-117 (No abstract available)
(3) ARRS Meeting 1996, Abstract #213
(4) AJR 1995; Apr 164(4): 831-835
(5) Radiol Clin North Am 1998; 36 (1): 29-55
(6) AJR 1999; Baque-Juston MC, et al. Pericardial thickening or effusion in patients with pulmonary artery hypertension: A CT study. 172: 361-364
(7) Radiology 1999; Remy-Jardin M, Remy J. Spiral CT angiography of the pulmonary circulation. 212: 615-636
(8) Thorax 1999; Peacock AJ. Primary pulmonary hypertension. 54: 1107-1118 (Review. No abstract available.)
(9) Radiographics 2000; Frazier AA, et al. From the archives of the AFIP. Pulmonary vasculature: Hypertension and infarction. 20: 491-524
(10) Chest 2000; Rich S, et al. Anorexigens and pulmonary hypertension in the United States. Results from the surveillance of North American pulmonary hypertension. 117: 870-874
(11) Chest 2000; Mehta NJ, et al. HIV-related pulmonary hypertension. Analytic review of 131 cases. 118: 1133-1141
(12) Radiology 2002; Bugnone AN, et al. Imaging findings in human immunodeficiency virus-related pulmonary hypertension: report of five cases and review of the literature. 223: 820-827
(13) J Nucl Med 2002; Fukuchi K, et al. Quantitative analysis of lung perfusion in patients with primary pulmonary hypertension. 43: 757-761
(14) Radiology 2002; Hansell DM, et al. Small-vessel diseases of the lung: CT-pathologic correlates. 225: 639-653
(15) J Nucl Cardiolo 2003; Boyce PD, et al. Pulmonary hypertension: work in progress. 10: 413-423
(16) AJR 2004; Resten A, et al. Pulmonary hypertension: CT of the chest in pulmonary venoocclusive disease. 183: 65-70
(17) Radiographics 2006; Restrepo CS, et al. Cardiovascular complications of human immunodeficiency virus infection. 26: 213-231
(18) Radiographics 2006; Castaner E, et al. Congenital and acquired pulmonary artery anomalies in the adult: radiologic overview. 26: 349-371
(19) J Nucl Med 2007; Tunariu N, et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic disease as a treatable cause of pulmonary hypertension. 48: 680-684
(20) Radiographics 2010; Grosse C, Grosse A. CT findings in diseases associated with pulmonary hypertension: a current review. 30: 1753-1777
(21) AJR 2011; Shehata ML, et al. Myocardial delayed enhancement
in pulmonary hypertension: pulmonary hemodynamics, right
ventricular function, and remodeling. 196: 87-94
(22) Radiographics 2012; Pena E, et al. Pulmonary hypertension:
how the radiologist can help. 32: 9-32
(23) Radiographics 2012; Capobianco J, et al. Thoracic
manifestations of collagen vascular diseases. 32: 33-50
(24) AJR 2012; Nakazono T, et al. HIV-related cardiac
complications: CT and MRI findings. 198: 364-369
(25) AJR 2012; Lichtenberger JP, et al. What a differetial a
virus makes: a practical approach to thoracic imaging findings in
the context of HIV infection- Part 2, extrapulmonary pulmonary
findings, chronic lung disease, and immune reconstitution
syndrome. 198: 1305-1312
(26) AJR 2012; Barbosa EJM, et al. Current role of imaging in the
diagnosis and management of pulmonary hypertension. 198: 1320-1331
(27) Radiographics 2014; Chou SHS, et al. Thoracic diseases
associated with HIV infection in the era of anti-retroviral
therapy: clinical and imaging findings. 34: 895-911
(28) AJR 2017; Grosse A, et al. Distinguishing chronic
thromboembolic pulmonary hypertension from other causes of
pulmonary hypertension using CT. 209: 1228-1238
(29) Radiographics 2018; Jaramillo FA, et al. Approach to
pulmonary hypertension: from CT to clinical diagnosis. 38: 357-373
(30) AJR 2018; Scelsi CL, et al. Egg-and-banana sign: a novel
diagnostic CT marker for pulmonary hypertension. 210: 1235-1239
(31) J Nucl Med 2019; Mehari A, et al. Abnormal
ventilation-perfusion scan is associated with pulmonary
hypertension in sickle cell adults. 60: 86-92
(32) Radiology 2020; Delhaye C, et al. Pulmonary veno-occlusive
disease and pulmonary capillary hemangiomatosis. 295: 240-244
(33) Radiographics 2020; Broncano J, et al. Cardiac MRI in
pulmonary hypertension: from magnet to bedside. 40: 982-1002
(34) Radiology 2021; Remy-Jardin M, et al. Imaging of pulmonary
hypertension in adults: a position paper from the Fleischner
society. 298: 531-549