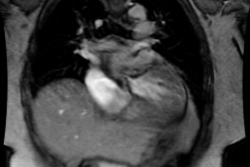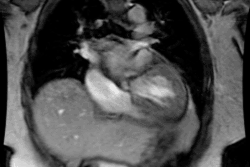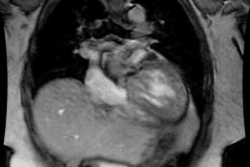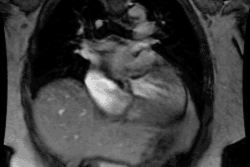
Cardiomyopathy:
Clinical:
Cardiomyopathies Can be classified as either hypertrophic, dilated, or restrictive.
Hypertrophic Cardiomyopathy:
Hypertrophic cardiomyopathy (HCM) is an inheritable genetic disorder (autosomal dominant mode of inheritance with variable expression in 50-60% of cases [8,20]) characterized by inappropriate left ventricular hypertrophy (greater than 15mm [11]) often with left ventricular outflow tract (LVOT) obstruction. Right ventricular involvement occurs in about 17-18% of cases- most commonly involving the mid to apical portion of the right ventricle [8,20]. There is often impaired regional myocardial function, arrhythmias, and decreased coronary flow reserve [7]. The condition is cause by mutations in one of the genes encoding sarcomere proteins which are composed of thick or thin filaments (more than 1500 different mutations have been identified) [16,17,18,27,28]. Beta-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) account for the majority of cases [27]. Screening of family members of a patient with HCM is important because the first-degree relatives of such a patient have a 50% chance of being a gene carrier (although expression is variable) [16]. The incidence in the United States is approximately one in 500 or 0.2%, of the population [17].
Most cases of HCM are phenotypically expressed in adolescence or
early adulthood [8]. There is notable heterogeneity in the
clinical expression, natural history, and prognosis [10]. Patients
are usually asymptomatic or mildly symptomatic [12]. However.
hypertrophic cardiomyopathy is the most common cardiac death in
young adults and is a well-recognized cause of sudden death in
athletes [23]. Apical HCM produces giant inverted anterolateral
T-waves on the ECG [24]. Chest pain is also a common symptom of
patients with HCM and it be the result of epicardial stenoses
(7-25%, particularly in elderly patients), small intramural
coronary artery dysplasia, increased demands of the hypertrophied
myocardium, and less commonly, due to bridging of the epicardial
coronary arteries (which can be seen in up to 41% of patients)
[25,26]. Unfortunately, chest pain symptoms are difficult to
evaluate in these patients as false positive myocardial perfusion
SPECT scans can occur in up to 63% [25].
Histologically there is muscular hyperplasia with myocardial
cellular disorganization, myocardial fiber disarray, and fibrosis
[11,16]. In about 3-9% of patients with HCM, the LV slowly (over
years) transitions from a hypertrophied chamber with preserved
systolic function, to a dilated, scarred chamber with thinned
walls and a reduced ejection fraction (end-stage HCM) [18,27,29].
Atrial fibrillation is among the most common complication of hypertrophic cardiomyopathy- occurring in over 20% of unselected patients and it usually implies an adverse prognosis due to an increased prevalence of heart failure-related complications [13]. The histologic changes (myocardial hypertrophy, myofiber disarry, and replacement fibrosis [27]) also result in an increased risk for ventricular dysrhythmia which is a common cause of mortality in these patients (HCM is a particularly common cause of sudden cardiac death in young people- particularly athletes [16,18]). Sudden cardiac death occurs in less than 1% (0.8%) of asymptomatic patients per year, but in up to 6% of patients with high risk factors such as LV maximal thickness of 30mm or more, LVOT gradient of 30 mm Hg or more at rest or 50 mm Hg or more with provocation, LV dilatation with depressed ejection fraction, the presence of fibrosis (greater extent associated with greater risk), perfusion defects, or reduced functional reserve flow [16,27]. Because there is a linear relationship between maximal myocardial thickness and risk for sudden cardiac death, a maximal myocardial thickness of 30 mm or more is an indication for ICD placement [23].
Most often the distribution of hypertrophy is asymmetric and
segmental, while diffuse concentric forms represent a minority of
cases (about 5%) [24]. However, other authors state that the
condition can also be symmetric (up to 42% of cases [8]),
midventricular, or mass-like [8,16]. In 70% of patients, the
hypertrophied regions involve the basal segments of the anterior
ventricular septum and anterior wall [24]. The two most recognized
distinct sub-types of hypertrophic cardiomyopathy- asymmetric
septal hypertrophy (ASH), and apical hypertrophy (apical
hypertrophy accounts for less than 10% of cases of HCM [24])
[8,16]. The usual diagnostic criterion for HCM is a maximal LV
wall thickness greater than or equal to 15 mm in the end-diastolic
phase [16]. Delayed myocardial enhancement is seen in 50-80% of
patients with HCM and is associated with nonsustained ventricular
tachycardia and sudden cardiac death [23].
An impairment in diastolic function (such as the peak filling
rate < 2.5 end-diastolic volumes and a time to peak filling
rate > 180 ms) is a characteristic of HCM (systolic function of
the heart is normal or even increased early in the disease)
[14,17,18,20,27]. The altered LV filling dynamics and reduced LV
distensibility are associated with a reduced LV stroke volume and
increased LV filling pressures [14]. Howeevr, the LV ejection
fraction is usually normal at the time of diagnosis [20]. In
patients with obstructive HCM a significant prolongation of LV
systole at the expense of diastolic time is seen at rest and with
exercise, the abnormal shortening of LV diastolic time is even
more greatly accentuated further limiting the time available for
myocardial perfusion [14].
Microvascular dysfunction is also a well-recognized feature of
hypertrophic cardiomyopathy [22]. The greater the myocardial wall
thickness, the greater the degree of impaired
diapyridamole-induced hyperemia and myocardial flow reserve on PET
13N-NH3 imaging [22]. Studies have also
found a higher prevalence of abnormal TID index (up to 55% of
patients) and blunted LVEF reserve in HCM patients [22].
In patients with HCM, abnormalities in 123I-BMIPP metabolism can be found in many LV segments with normal resting perfusion- particularly the most hypertrophied [18]. The abnormal BMIPP uptake is often most pronounced in the subendocardial regions [18]. The degree of regional LV BMIPP uptake has been shown to directly correlate with the peak negative myocardial velocity gradient measured by tissue Doppler [18]. Followup of asymptomatic HCM patients has shwon that abnormal BMIPP uptake at baseline can predict future deterioration in LV function [18].
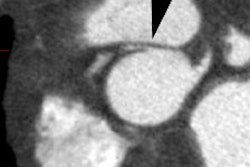
a) Asymmetric septal hypertrophy (ASH):
ASH is the most common hypertrophic cardiomyopathy (60-70% of
cases [16]) and is characterized by a disproportionate thickening
of the interventricular septum (as compared to the posterolateral
wall or apex) which results in a sub-valvular left ventricular
outflow tract (LVOT) obstruction in 20-30% of cases [8,28]. The
disorder is characterized by decreased systolic thickening of the
septum, abnormal anterior motion of the anterior leaflet of the
mitral valve during systole- referred to as SAM (due to venturi or
drag forces [8]). The degree of LVOT obstruction is exacerbated by
the systolic anterior motion of the mitral valve. Over time, the
repeated mitral valve impact on the septum leads to
thickening of the anterior leaflet (fibrosis) and chordae with
subsequent mitral regurge [8]. The regurgitation due to SAM is
typically directed toward the posterior wall of the LA, forming a
right angle with the turbulent LVOT flow, and occurs in mid to
late systole [28]. The anterolateral wall is also commonly
involved by the disorder.
Patients may be asymptomatic or may present with dyspnea,
presyncope, syncope, or angina [8]. LVOT obstruction is defined by
instantaneous peak gradients ≥ 30 mmHg at rest measured with
continuous-wave Doppler echocardiography [27,28]. Patients
with LVOT obstruction and severe symptoms unresponsive to medical
therapy (about 5% of patients) are candidates for surgical
myomectomy or septal alcohol ablation [9,16]. Patients with a
dynamic obstruction (peak instantaneous gradient ≥ 50 mm Hg at rest or with provocation)
or a septal thickness of more than 1.6 cm should also be
considered for septal reduction therapy (surgical myomectomy or
transcatheter alcohol ablation) [16,27]. Surgical resection of
the hypertrophic segment of the septum has an initial success
rate of 90% in decreasing symptoms and LVOT obstruction [16]. In
about 70% of patients, the lessening of symptoms is maintained
[16]. Septal alcohol ablation (producing iatrogenic
infarction of the basal interventricular septum) has a higher
complication rate [9].
The normal thickness of the LV is 11 mm or less, measured during
diastole [28]. The diagnosis of asymmetric septal hypertrophy is
made when the septal thickness is greater or equal to 15 mm or
when the ratio of the septal thickness to the thickness of the
inferior wall at the mid-ventricular level is greater than 1.5
[28].
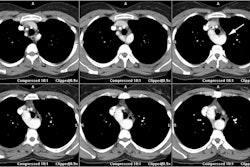
On spin echo MR images findings include asymmetric thickening of the ventricular septum and regions of decrease signal intensity (which may represent areas of fibrosis). Asymmetric septal HCM is diagnosed when the septal thickness is greater than or equal to 15mm or when the ratio of the septal thickness to the thickness of the inferior wall of the LV is greater than 1.5 at the midventricular level [16] (other authors indcaited a ratio of 1.3 or more [23]).
Cine MR imaging can demonstrate the lack of change in septal thickness between systole and diastole, regurgitant abnormalities associated with septal hypertrophy (due to paradoxical anterior motion of the anterior mitral valve leaflet during systole which produces mitral regurgitation).
On contrast enhanced MR, delayed enhancement of the left ventricular myocardium can be seen in up to 80% of patients [8]. The delayed enhancement is usually patchy or a speckled pattern, involves the mid portion of the myocardium, frequently involves the areas of myocardial thickening, and most commonly occurs in the septum (particularly at sites of attachment of the RV to the LV) [6,8,17,29]. Hyperenhancement on delayed enhanced MR imaging has been correlated with regions of fibrosis histologically [28]. In patients with HCM, the presence of delayed enhancement on contrast MR imaging and the number of involved segments correlates with the presence of ventricular techycardia [9,18]. More extensive enhancement is also found in patients with more clinical risk factors for sudden cardiac death [9]. The presence of absence of delayed myocardial enhancement is the primary predictor of nonsustained ventricular tachycardia [17].
b) Symmetric HCM:
Symmetric/concentric HCM is characterized by diffuse thickening
of the LV [16,28]. It is the second most common phenotype and may
account for up to 42% of cases of HCM [16,28]. The diagnosis
should be made when there are no secondary causes of hypertrophy
such as hypertension or aortic stenosis, as well as other causes
of concentric LV thickening such as amyloidosis [28]. Areas of
late myocardial enhancement on MR imaging has been associated with
an increased risk for arrhythmia, sudden cardiac death, and
implantable cardiac defibrillator discharge [20].
In patients with increased LV thickness from an athletic heart
there is preservation of the LV cavity size of more than 55 mm and
there is no delayed enhancement on post gadolinium imaging [28].
Additionally, in athletes, there is a 2-5 mm regression in LV wall
thickness over a short deconditioning period of about 3 months
which is not observed in HCM patients [28]. In HTN the LV wall
thickness rarely exceeds 16 mm [28].
c) Apical hypertrophy (Yamaguchi syndrome):
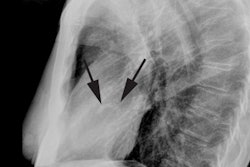
In this rare disorder, there is marked myocardial thickening to the left ventricular apex which produces a characteristic "spade-shaped" left ventricle on ventriculography [5]. Apical HCM accounts for up to 15-25% of cases of HCM in Japan, but only about 1-2% of cases in Western countries [8,15,28]. The disorder is usually clinically benign and frequently associated with systemic hypertension in middle aged to older men [16]. Patients may present with exertional angina or dyspnea and may have ECG findings similar to those in acute coronary syndromes [28]. The condition has a relatively good prognosis and is rarely associated with sudden cardiac death [16], but up to one-third of patients may present with a severe complication such as MI, atrial fibrillation, and stroke [28]. A characteristic ECG change associated with the disorder is LVH by voltage criteria and giant inverted T-waves (>10 mm) in the lateral precordial leads (seen in up to 50% of cases) [5,8,15,21]. There may be a genetic basis for the disorder as it is encountered much more frequently in Asian populations, but rare in Western countries [21]. The etiology is likely multifactoral and related to impaired coronary flow reserve, a decreased capillary myocardial ratio, inadequate diastolic relaxation, and compression of the coronary vessels [5]. Despite the lack of obstructive coronary artery disease, apical infarcts have been reported in up to 10% of patients [5]. Concomitant apical involvement of the RV is also commonly seen [8]. When referred for myocardial perfusion imaging, patients will demonstrate a characteristic "solar polar" pattern on quantitative polar map- with an intensely bright area in the apical segment surrounded by a circumferential ring of decreasing counts [15].
On perfusion imaging, rest images typically show pronounced tracer uptake in the LV apex [5]. A characteristic "solar polar" map has been described with a central focus of increased apical activity surrounded by a ring of decreasing counts [5]. Perfusion abnormalities consistent with ischemia are commonly seen- particularly in the regions of prominent hypertrophy [5].
Diagnostic criteria for apical HCM include an absolute apical
wall thickness of greater than 15 mm or a ratio comparing apical
left ventricular and basal LV wall thickness of ≥ 1.3-1.5 [8].
Subjective criteria include obliteration of the LV apical cavity
in systole and failure to identify a normal progressive reduction
in LV wall thickness toward the apex [8]. On vertical long axis
views there is a "spade-like" configuration of the LV cavity at
end diastole [16,28]. The apical portion of the RV is often
involved as well [28]. On contrast enhanced MR imaging, there is
scattered, patchy enhancement within the areas of hypertrophied
myocardium [7]. The degree of enhancement is inversely correlated
with regional contraction [7].
d) Midventricular hypertrophy:
Isolated midventricular HCM is a rare disorder which produces a
characteristic hourglass or dumbbell shape to the LV [28].
Midsystolic muscular apposition of the interventricular septum
with the free wall of the LV can lead to mid-cavity obstruction
defined as a midcavity gradient greater than 30 mm Hg [28].
Systolic anterior motion of the mitral valve and LVOT obstruction
are typically absent [28]. The pressure overload in the apical LV
chamber increases myocardial stress and decreases perfusion,
leading to fibrosis and development of an apical aneurysm (in up
to 28% of patients) [28].
Dilated Cardiomyopathy:
Dilated cardiomyopathy is a pathophysiologic classification
characterized by ventricular dilatation and systolic dysfunction
with decreased ejection fraction. Dilated cardiomyopathy is the
most common of the three forms of cardiomyopathy [6]. Although it
may involve the right ventricle, the left ventricle is usually
more severely involved and hence, more dilated (although four
chamber dilatation is not uncommon). Histologically, DCM is
characterized by progressive interstitial fibrosis and myocyte
degeneration- and unlike CAD, there is mid wall rather than
subendocardial involvement [11]. Patients usually present with
progressive dyspnea and orthopnea [20].
On contrast enhanced MR imaging, up to 59% of patients will not show abnormal enhancement [7,29]. When seen, delayed enhancement on MR imaging can have two patterns- patchy with longitudinal mid wall enhancement that spares the subendocardium and subepicardium (more common- 28-35% of patients [29]); or subendocardial or transmural enhancement that is indistinguishable from ischemic heart disease (less common) [6]. The presence of mid-wall fibrosis in DCM is associated with an increased risk of sudden cardiac death and ventricular tachycardia (independent of other more traditional makers of increased risk such as LVEF) [11].
Etiologies of dilated cardiomyopathy include:
- Idiopathic: The most common form of dilated cardiomyopathy is idopathic (50%) [20]. As many as 35% of all cases of idiopathic dilated cardiomyopathy are familial or inherited, with autosomal dominant, autosomal recessive, maternal, or X chromosome-linked traits [23]. Idiopathic dilated cardiomyopathy is generally associated with a poor prognosis with a five-year mortality between 20% to 50% [4]. Effective early therapeutic intervention can result in an improved prognosis [4].
- Coronary artery disease
- Post-infectious (viral): A pathogenic link exists between infectious agents (usually viral) and subsequent immune mediated myocardial damage [3]. About 21% of patients with a clinical or histologic diagnosis of acute myocarditis (viral or of unknown etiology) will progress to a dilated cardiomyopathy over a mean of 3 years [3].
- HIV infection: Dilated cardiomyopathy occurs with a prevalence
of 8-30% in HIV patients [19]. It is usually seen in patients
with advanced immunosuppression and low CD4 counts [19].
- Post partum: Peripartum cardiomyopathy is rare [20]. It is
characterized by the onset of heart failure in the last month of
pregnancy or in the first 5 months postpartum without previous
underlying heart disease [20].
- Familial
- Toxic agents:
Chemotherapeutic agents. Adriamycin (doxorubicin) is a cardiotoxic chemotherapeutic agent [2]. Acute cardiac toxicity from the agent is uncommon, transient, and should improve after about 1 week. Chronic adriamycin cardiomyopathy is generally noted to occur approximately 30 days, up to 6 months, after the last dose. Cardiac toxicity is dose dependent and is rarely seen at doses less than 400 mg/m2. Pre-existing conditions such as age greater than 70 years, underlying coronary artery disease, and adjuvant chemotherapy increase the risk for developing cardiotoxicity. Cardioprotectants such as dexrazoxane have been used to attenuate the effects of adriamycin on the heart [2].
Alcohol
Restrictive Cardiomyopathy:
Restrictive cardiomyopathy is also a pathophysiologic classification characterized by impairment of ventricular diastolic filling and diastolic volume [12]. Restrictive cardiomyopathies produce decreased left ventricular compliance which results in resistance to left atrial emptying and poor diastolic filling. Restrictive cardiomyopathy can be idiopathic or secondary to an infiltrative or storage diseases [12]. It can also be associated with myocardial disorders such as hypereosinophilic syndrome [12]. Etiologies include:
1- Sarcoid or amyloid - which produce increased left ventricular thickness associated with heterogeneous signal characteristics on MR imaging.
2- Hemochromotosis also produces a restrictive cardiomyopathy which is characterized by signal loss of the myocardium secondary to iron deposition. Patients with thalassemia major also suffer from iron overload cardiomyopathy [11].
3- Radiation fibrosis can also produce a restrictive cardiomyopathy [6].
Hemodynamic and clinical features of this disorder are difficult to distinguish from constrictive pericarditis. The pericardium should be of normal thickness in patients with restrictive cardiomyopathy, while a pericardial thickness of greater than 4 mm is typical of constrictive pericarditis [12]. On MR, in restrictive cardiomyopathy, septal convexity is maintained in all respiratory phases, whereas in constrictive pericarditis, septal flatenning can be observed in early inspiration [12].
REFERENCES:
(1) Magn Reson Imaging Clin N Am 1996; May 4(2): 269-286
(2) J Thorac Imag 1999; Conces DJ. Noninfectious lung disease in immunocompromised patients. 14: 9-24
(3) J Nucl Med 2002; Merlet P, et al. Myocardial adrenergic dysinnervation in dilated cardiomyopathy: cornerstone or epiphenomenon? 43: 536-539
(4) J Nucl Cardiol 2003; Kasama S, et al. Myocardial contractile reserve determined by dobutamine stress Tc-99m tetrofosmin quantitative gated SPECT predicts late spontaneous improvement in cardiac function in patients with recent-onset dilated cardiomyopathy. 10: 607-614
(5) J Nucl Cardiol 2005; Postischemic stunning in apical hypertrophic cardiomyopathy. 12: 231-233
(6) AJR 2007; Lim RP, et al. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. 188: 1675-1681
(7) Radiographics 2006; Vogel-Claussen J, et al. Delayed enhancement MR imaging: utility in myocardial assessement. 26: 795-810
(8) AJR 2007; Hansen MW, Merchant N. MRI of hypertrophic cardiomyopathy: Part I, MRI appearances. 189: 1335-1343
(9) AJR 2007; Hansen MW, Merchant N. MRI of hypertrophic cardiomyopathy: Part 2, differential diagnosis, risk stratification, and post treatment MRI appearances. 189: 1344-1352
(10) J Nucl Med 2008; Sotgia B, et al. Spatial relationship between coronary microvascular dysfunction and delayed contrast enhancement in patients with hypertrophic cardiomyopathy. 49: 1090-1096
(11) J Nucl Cardiol 2008; O'Hanlon R, et al. Evaluation of non-ischemic cardiomyopathy using cardiovascular magnetic resonance. 15: 400-416
(12) AJR 2008; Belloni E, et al. MRI of cardiomyopathy. 191: 1702-1710
(13) J Nucl Cardiol 2009; Sciagra R, et al. Relationship between atrial fibrillation and blunted hyperemic myocardial blood flow in patients with hypertrophic cardiomyopathy. 16: 92-96
(14) J Nucl Cardiol 2009; Plehn G, et al. Effects of exercise on the duration of diastole and on interventricular phase differences in patients with hypertrophic cardiomyopathy: relationship to cardiac output reserve. 16: 233-243
(15) J Nucl Cardiol 2009; Cianciulli T F, et al. Myocardial perfusion SPECT in the diagnosis of apical hypertrophic cardiomyopathy. 16: 391-395
(16) Radiographics 2010; Chun EJ, et al. Hypertrophic cardiomyopathy: assessment with MR imaging and multi-detector CT. 30: 1309-1328
(17) AJR 2010; Bluemke DA. MRI of nonischemic cardiomyopathy. 195: 935-940
(18) J Nucl Cardiol 2011; Shirani J, Dilsizian V. Nuclear cardiac
imaging in hypertrophic cardiomyopathy. 18: 123-134
(19) AJR 2012; Nakazono T, et al. HIV-related cardiac
complications: CT and MRI findings. 198: 364-369
(20) Radiology 2012; O'Donnell DH, et al. Cardiac MR imaging of
nonischemic cardiomyopathies: imaging protocols and spectra of
appearances. 262: 403-422
(21) J Nucl Cardiol 2012; Hsieh BPC, Travin MI. Myocardial
perfusion imaging findings in apical hypertrophic cardiomyopathy.
19: 172-176
(22) J Nucl Med 2012; Bravo PE, et al. PET/CT assessment of
symptomatic individuals with obstructive and nonobstructive
hypertrophic cardiomyopathy. 53: 407-414
(23) Radiographics 2013; Stojanovska J, et al. Congenital and
hereditary causes of sudden cardiac death in young adults:
diagnosis, differential diagnosis, and risk stratification. 33:
1977-2011
(24) Radiology 2014; Bogaert J, Olivotto I. MR imaging in
hypertrophic cardiomyopathy: from magnet to bedside. 273: 329-348
(25) J Cardiovasc Compt Tomogr 2014; Shariat M, et al. Utility of
conary CT angiography in outpatients with hypertrophic
cardiomyopathy presenting with angina symptoms. 8: 429-437
(26) J Cardiovasc Comput Tomogr 2014; Blankstein R, Rowin EJ.
What is the best imaging test for patients with hypertrophic
cardiomyopathy? It depends on the clinical question! 8: 438-441
(27) J Nucl Cardiol 2015; Delgado V, et al. Clinical topic:
nuclear imaging in hypertrophic cardiomyopathy. 22: 408-418
(28) Radiographics 2016; Baxi AJ, et al. Hypertrophic
cardiomyopathy from A to Z: genetics, pathophysiology, imaging,
and management. 36: 335-354
(29) Radiographics 2017; Hashimura H, et al.
Radiologic-pathologic correlation of primary and secondary
cardiomyopathies: MR imaging and histopathologic findings in
hearts from autopsy and transplantation. 37: 719-736